Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:1
-
Sprint:Spring 1 2022 Jan 3 - Jan 14, Spring 2 2022 Jan 18 - Jan 28
Description
Previous analysis of the RNA-Seq data found that the treatments triggered changes in expression and splicing. Since our first processing of the data, we added a new dataset in which a methylation inhibitor was applied, but this dataset has not been processed as yet.
A new RNA-Seq data analysis pipeline has been developed that uses the Nextflow workflow system, and we've been using this workflow system to process data from the pollen heat stress project. Also, this workflow management system is better equipped to accommodate diverse samples, e.g., sample libraries sequenced using different strategies (single- versus paired-end) and read lengths.
Since we need to process a new methylation-inhibitor RNA-Seq dataset and incorporate it into our analysis, let's reprocess the data using a more up-to-date workflow - the nc-core/rnaseq pipeline.
The first steps in doing this will be to:
- generate a comma-separated sample sheet data file that relates SRA run identifiers to experimental attributes, required for running nextflow. Note that we can also use the sample sheet as inputs for statistical analyses.
- generate a script that will download the SRA data files and convert them to fastq, required for running the pipeline. (Note: The project identifier is: PRJNA481973/)
The sample sheet data file columns will include the following fields:
- SRA run identifier (e.g, SRR7591232)
- fastq_1 - SRR name with _1 appended (e.g, SRR7591232_1)
- fastq_2 - SRR name with _2 appended, or blank for single-end samples ( (e.g, SRR7591232_2)
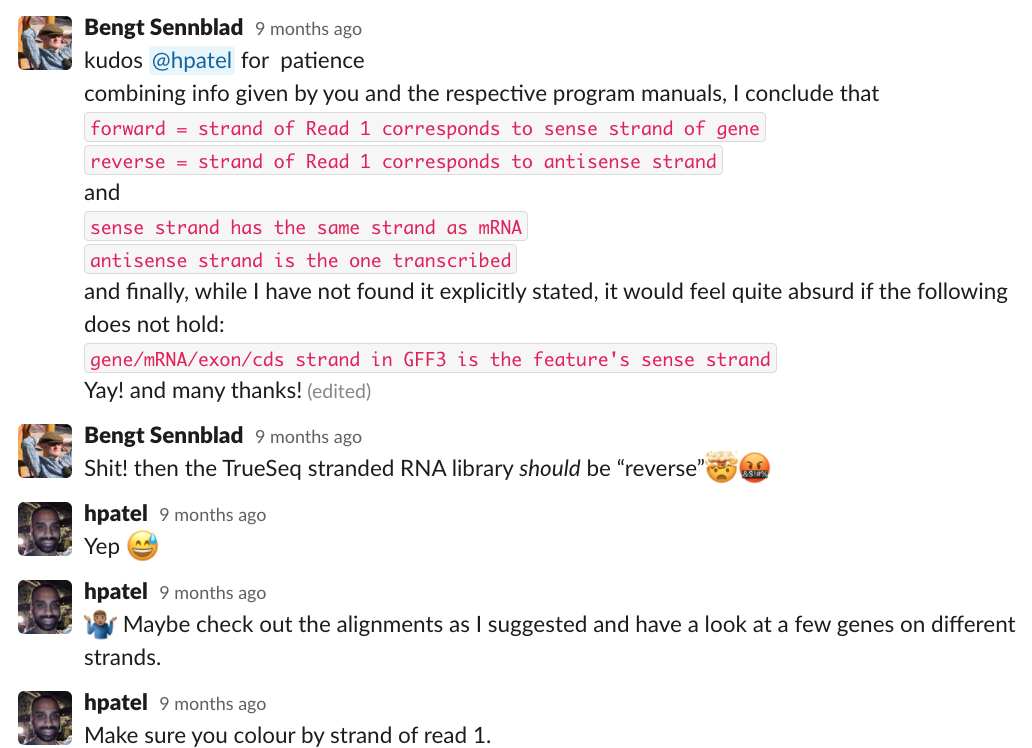
- strandedness - should be "reverse" for Truseq Illumina protocol (see attached image from nf-core/rnaseq slack)
- genotype - A (Agami), M (M103)
- treatment - C (control), E (salt)
- 5-Azacytidine treatment - Y (treated), N (not treated)
- tissue - S (shoot), R (root)
- replicate - 1, 2, 3
- read length
Attachments
Issue Links
Activity
| Field | Original Value | New Value |
|---|---|---|
| Epic Link | IGBF-3039 [ 21553 ] |
| Rank | Ranked higher |
| Attachment | sra_explorer_metadata.tsv [ 17043 ] |
Used cut, uniq, and sort to retrieve the 29 SRR identifiers, in order, for the RNA-Seq datasets listed in the attached tsv file.
Using faster-dump, a utility part of SRA tools (released from NCBI), we can retrieve data using the following bash commands.
Dividing into 6 sections of consecutive numbers, with bash commands to run:
fasterq-dump -S -P -p SRR759123{2..4}
SRR7591232
SRR7591233
SRR7591234
fasterq-dump -S -P -p SRR759124{3..5}
SRR7591243
SRR7591244
SRR7591245
fasterq-dump -S -P -p SRR7591249
SRR7591249
fasterq-dump -S -P -p SRR75912{58..66}
SRR7591258
SRR7591259
SRR7591260
SRR7591261
SRR7591262
SRR7591263
SRR7591264
SRR7591265
SRR7591266
fasterq-dump -S -P -p SRR75912{69..73}
SRR7591269
SRR7591270
SRR7591271
SRR7591272
SRR7591273
fasterq-dump -S -P -p SRR759128{2..9}
SRR7591282
SRR7591283
SRR7591284
SRR7591285
SRR7591286
SRR7591287
SRR7591288
SRR7591289
Nowlan Freese - is the above information enough to enable you to create a first draft of the sample sheet file?
| Description |
Create a comma-separated data file that relates SRA run identifiers to experimental attributes.
Ensure that the sample sheet can be used to run the Nextflow rna-seq pipeline. Project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/] The columns should include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended # fastq_2 - SRR name with _2 appended, or blank for single-end samples # strandedness (need to look up how Nextflow designates this) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-aza-2’-deoxycytidine (5-azaC) treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
Create a comma-separated data file that relates SRA run identifiers to experimental attributes.
Ensure that the sample sheet can be used to run the Nextflow rna-seq pipeline. Note that we can also use the sample sheet as inputs for statistical analyses. Project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/] The columns should include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended # fastq_2 - SRR name with _2 appended, or blank for single-end samples # strandedness (need to look up how Nextflow designates this) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-aza-2’-deoxycytidine (5-azaC) treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
| Description |
Create a comma-separated data file that relates SRA run identifiers to experimental attributes.
Ensure that the sample sheet can be used to run the Nextflow rna-seq pipeline. Note that we can also use the sample sheet as inputs for statistical analyses. Project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/] The columns should include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended # fastq_2 - SRR name with _2 appended, or blank for single-end samples # strandedness (need to look up how Nextflow designates this) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-aza-2’-deoxycytidine (5-azaC) treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
Previous analysis of the RNA-Seq data found that the treatments triggered changes in expression and splicing. Since our first processing of the data, we added a new dataset in which a methylation inhibitor was applied, but this dataset has not been processed as yet.
A new RNA-Seq data analysis pipeline has been developed that uses the Nextflow workflow system, and we've been using this workflow system to process data from the pollen heat stress project. Also, this workflow management system is better equipped to accommodate diverse samples, e.g., sample libraries sequenced using different strategies (single- versus paired-end) and read lengths. Since we need to process a new methylation-inhibitor RNA-Seq dataset and incorporate it into our analysis, let's reprocess the data using a more up-to-date workflow - the nc-core/rnaseq pipeline. The first steps in doing this will be to: * generate a comma-separated sample sheet data file that relates SRA run identifiers to experimental attributes, required for running nextflow. Note that we can also use the sample sheet as inputs for statistical analyses. * generate a script that will download the SRA data files and convert them to fastq, required for running the pipeline. (Note: The project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/]) The sample sheet data file columns will include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended (e.g, SRR7591232_1) # fastq_2 - SRR name with _2 appended, or blank for single-end samples ( (e.g, SRR7591232_2) # strandedness (need to look up how Nextflow designates this) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-aza-2’-deoxycytidine (5-azaC) treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
| Comment | [ The Bioconductor and probably other libraries as well in R can probably assemble this information automatically. ] |
| Attachment | strand.png [ 17044 ] |
| Description |
Previous analysis of the RNA-Seq data found that the treatments triggered changes in expression and splicing. Since our first processing of the data, we added a new dataset in which a methylation inhibitor was applied, but this dataset has not been processed as yet.
A new RNA-Seq data analysis pipeline has been developed that uses the Nextflow workflow system, and we've been using this workflow system to process data from the pollen heat stress project. Also, this workflow management system is better equipped to accommodate diverse samples, e.g., sample libraries sequenced using different strategies (single- versus paired-end) and read lengths. Since we need to process a new methylation-inhibitor RNA-Seq dataset and incorporate it into our analysis, let's reprocess the data using a more up-to-date workflow - the nc-core/rnaseq pipeline. The first steps in doing this will be to: * generate a comma-separated sample sheet data file that relates SRA run identifiers to experimental attributes, required for running nextflow. Note that we can also use the sample sheet as inputs for statistical analyses. * generate a script that will download the SRA data files and convert them to fastq, required for running the pipeline. (Note: The project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/]) The sample sheet data file columns will include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended (e.g, SRR7591232_1) # fastq_2 - SRR name with _2 appended, or blank for single-end samples ( (e.g, SRR7591232_2) # strandedness (need to look up how Nextflow designates this) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-aza-2’-deoxycytidine (5-azaC) treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
Previous analysis of the RNA-Seq data found that the treatments triggered changes in expression and splicing. Since our first processing of the data, we added a new dataset in which a methylation inhibitor was applied, but this dataset has not been processed as yet.
A new RNA-Seq data analysis pipeline has been developed that uses the Nextflow workflow system, and we've been using this workflow system to process data from the pollen heat stress project. Also, this workflow management system is better equipped to accommodate diverse samples, e.g., sample libraries sequenced using different strategies (single- versus paired-end) and read lengths. Since we need to process a new methylation-inhibitor RNA-Seq dataset and incorporate it into our analysis, let's reprocess the data using a more up-to-date workflow - the nc-core/rnaseq pipeline. The first steps in doing this will be to: * generate a comma-separated sample sheet data file that relates SRA run identifiers to experimental attributes, required for running nextflow. Note that we can also use the sample sheet as inputs for statistical analyses. * generate a script that will download the SRA data files and convert them to fastq, required for running the pipeline. (Note: The project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/]) The sample sheet data file columns will include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended (e.g, SRR7591232_1) # fastq_2 - SRR name with _2 appended, or blank for single-end samples ( (e.g, SRR7591232_2) # strandedness - should be "reverse" for Truseq Illumina protocol (see attached image from nf-core/rnaseq slack) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-aza-2’-deoxycytidine (5-azaC) treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
| Sprint | Spring 1 2022 Jan 3 - Jan 14 [ 136 ] |
| Rank | Ranked lower |
| Status | To-Do [ 10305 ] | In Progress [ 3 ] |
| Assignee | Nowlan Freese [ nfreese ] |
| Description |
Previous analysis of the RNA-Seq data found that the treatments triggered changes in expression and splicing. Since our first processing of the data, we added a new dataset in which a methylation inhibitor was applied, but this dataset has not been processed as yet.
A new RNA-Seq data analysis pipeline has been developed that uses the Nextflow workflow system, and we've been using this workflow system to process data from the pollen heat stress project. Also, this workflow management system is better equipped to accommodate diverse samples, e.g., sample libraries sequenced using different strategies (single- versus paired-end) and read lengths. Since we need to process a new methylation-inhibitor RNA-Seq dataset and incorporate it into our analysis, let's reprocess the data using a more up-to-date workflow - the nc-core/rnaseq pipeline. The first steps in doing this will be to: * generate a comma-separated sample sheet data file that relates SRA run identifiers to experimental attributes, required for running nextflow. Note that we can also use the sample sheet as inputs for statistical analyses. * generate a script that will download the SRA data files and convert them to fastq, required for running the pipeline. (Note: The project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/]) The sample sheet data file columns will include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended (e.g, SRR7591232_1) # fastq_2 - SRR name with _2 appended, or blank for single-end samples ( (e.g, SRR7591232_2) # strandedness - should be "reverse" for Truseq Illumina protocol (see attached image from nf-core/rnaseq slack) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-aza-2’-deoxycytidine (5-azaC) treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
Previous analysis of the RNA-Seq data found that the treatments triggered changes in expression and splicing. Since our first processing of the data, we added a new dataset in which a methylation inhibitor was applied, but this dataset has not been processed as yet.
A new RNA-Seq data analysis pipeline has been developed that uses the Nextflow workflow system, and we've been using this workflow system to process data from the pollen heat stress project. Also, this workflow management system is better equipped to accommodate diverse samples, e.g., sample libraries sequenced using different strategies (single- versus paired-end) and read lengths. Since we need to process a new methylation-inhibitor RNA-Seq dataset and incorporate it into our analysis, let's reprocess the data using a more up-to-date workflow - the nc-core/rnaseq pipeline. The first steps in doing this will be to: * generate a comma-separated sample sheet data file that relates SRA run identifiers to experimental attributes, required for running nextflow. Note that we can also use the sample sheet as inputs for statistical analyses. * generate a script that will download the SRA data files and convert them to fastq, required for running the pipeline. (Note: The project identifier is: [PRJNA481973/|https://www.ncbi.nlm.nih.gov/bioproject/PRJNA481973/]) The sample sheet data file columns will include the following fields: # SRA run identifier (e.g, SRR7591232) # fastq_1 - SRR name with _1 appended (e.g, SRR7591232_1) # fastq_2 - SRR name with _2 appended, or blank for single-end samples ( (e.g, SRR7591232_2) # strandedness - should be "reverse" for Truseq Illumina protocol (see attached image from nf-core/rnaseq slack) # genotype - A (Agami), M (M103) # treatment - C (control), E (salt) # 5-Azacytidine treatment - Y (treated), N (not treated) # tissue - S (shoot), R (root) # replicate - 1, 2, 3 # read length |
| Attachment | samplesheet_RNA-Seq.csv [ 17050 ] |
I have attached the RNA-Seq sample sheet (samplesheet_RNA-Seq.csv).
Note that the file has a header line.
A few additional notes:
- Not all samples were sequenced with paired end (we did not pay for paired end, but some samples were placed on flow cells that were running paired end samples for other clients).
- The Agami salt treated Az treated shoot samples only had two biological replicates (2 and 3). My recollection is that something had happened to the biological replicate 1 sample during library preparation and we were unable to sequence it.
- Some samples (the Az samples) were sequenced multiple times. These samples have paired end reads and an additional single end sequencing run that was done on a separate date. All of the fastq files were submitted to the SRA (the initial paired fastq files as well as the additional single end fastq file). However, it is unclear to me how those files are stored by the SRA. It could be that the SRA has combined the two R1 fastq submissions, and this is why there is a discrepancy in the spot lengths (see comment below). We may need to look at this more closely when aligning the Az samples.
- The following samples had abnormal average spot lengths according to the SRA Run Selector: SRR7591233, SRR7591234, SRR7591249, SRR7591266, SRR7591286. These samples are the Az treated samples and are the same samples that were resequenced. This may be due to an issue with the sequencing itself or it may be due to the SRA combining R1 fastq files from two files (see above comment).
- I double-checked the forward/reverse logic and it does appear correct that these samples would be labeled as reverse for strandedness.
- The read lengths all appear to be 125bp.
| Assignee | Nowlan Freese [ nfreese ] | Ann Loraine [ aloraine ] |
| Status | In Progress [ 3 ] | To-Do [ 10305 ] |
| Status | To-Do [ 10305 ] | In Progress [ 3 ] |
| Status | In Progress [ 3 ] | Needs 1st Level Review [ 10005 ] |
| Sprint | Spring 1 2022 Jan 3 - Jan 14 [ 136 ] | Spring 1 2022 Jan 3 - Jan 14, Spring 2 2022 Jan 18 - Jan 28 [ 136, 137 ] |
| Rank | Ranked higher |
Adding sample sheet to the repository. Moving to Done.
| Status | Needs 1st Level Review [ 10005 ] | First Level Review in Progress [ 10301 ] |
| Status | First Level Review in Progress [ 10301 ] | Ready for Pull Request [ 10304 ] |
| Status | Ready for Pull Request [ 10304 ] | Pull Request Submitted [ 10101 ] |
| Status | Pull Request Submitted [ 10101 ] | Reviewing Pull Request [ 10303 ] |
| Status | Reviewing Pull Request [ 10303 ] | Merged Needs Testing [ 10002 ] |
| Status | Merged Needs Testing [ 10002 ] | Post-merge Testing In Progress [ 10003 ] |
| Resolution | Done [ 10000 ] | |
| Status | Post-merge Testing In Progress [ 10003 ] | Closed [ 6 ] |
| Assignee | Ann Loraine [ aloraine ] | Nowlan Freese [ nfreese ] |
Note that the sample sheet attached to this ticket is outdated. The newest version of the sample sheet can be found in the bseq_rice repository.
Used https://sra-explorer.info/ (SRA Explorer) to get tsv file (attached) listing SRR numbers for RNA-Seq data files associated with the experiment.
Next, we can use fasterq-dump (installed on the Charlotte HPC system) to download the files from the SRA and simultaneously convert them to fastq format.
See: this comment from
IGBF-2984"Investigate tools for detecting strandedness in RNA-Seq" for the correct options to use.