Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:0.5
-
Epic Link:
-
Sprint:Spring 2 2023 Jan 16
Description
Run nextflow for the dataset in:
/projects/tomato_genome/rnaseq/mark-2022-timeseries/30-771363348/00_fastq
This is the "time course" dataset discussed by Rasha at the 2023-01-17 group meeting. Note that she has already run nextflow for this dataset but using "unstranded" for the "strandedness" parameter in the "samples.csv" file. It turns out this dataset comes from libraries that were created using a strand-specific RNA-Seq library. To be on the safe side, we should re-run the pipeline using parameter "reverse", as indicated in the multiQC report included with Rasha's initial run of the nextflow nf-core rnaseq pipeline.
Kindly run the nf-core pipeline in this location:
- /nobackup/tomato_genome/mark-2022-timeseries
Note on attached files:
- multiqc report on the entire run done by Rasha is attached, copied from google drive location GTTR-NSF PGRP - 2020-24 IOS-1939255 > Experiments > Rasha_RNA-seq_Time_Course > Results > multiqc > star_salmon > multiqc_report.html
- Link: https://drive.google.com/drive/u/1/folders/1GJnZefP-7TE-ch-c0lblGZMOqpwSgZRK
- 2023-01-18_timeseries_multiqc_report.html - MultiQC report from re-running nextflow (Molly's new work)\
- sample.csv - new samples file used to re-run nextflow (Molly's new work)
Attachments
Issue Links
Activity
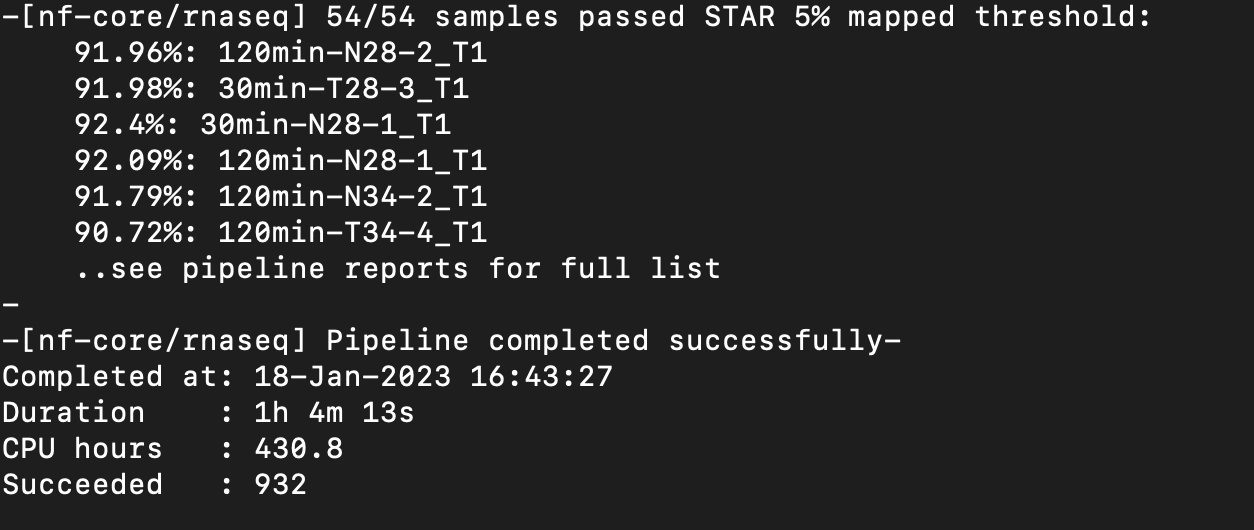
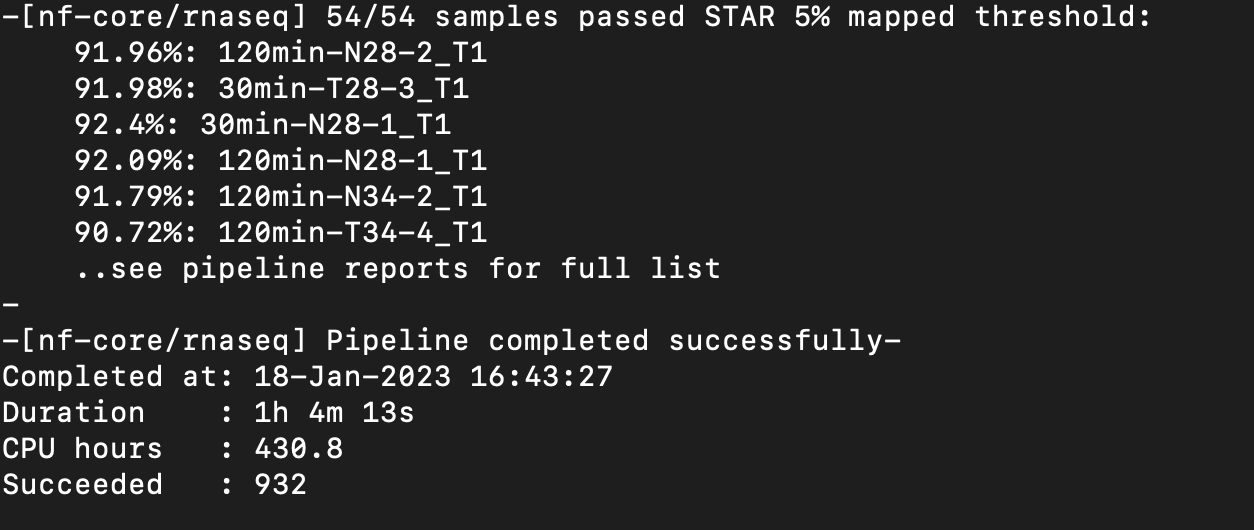
Nextflow Pipeline Ran Successfully!
Directory: /nobackup/tomato_genome/mark-2022-timeseries
Next steps: Rename sorted bam files and make scaled coverage graphs.
Scaled coverage graphs have been made and are located:
/nobackup/tomato_genome/mark-2022-timeseries/results/star_salmon
Notes: I can move the coverage graphs to their own directory if you would like. Let me know!
Multiqc report:
scp mdavi258@hpc.uncc.edu:/nobackup/tomato_genome/mark-2022-timeseries/results/multiqc/star_salmon/multiqc_report.html timeseries_multiqc_report.html
[^timeseries_multiqc_report.html]
Notes: Multiqc report seems to show better mapping and correct strandedness now compared to the previous report and nextflow run.
Next step: Pipeline, coverage graphs, and Multiqc report need to be reviewed.
[~aloraine]
I reviewed multiqc report and noticed no problems.
I migrated coverage graphs and bam files to igb quickload host and updated makeAnnotsXml.py in https://bitbucket.org/hotpollen/splicing-analysis/src/main/ to use the new files.
See: ManageQuickload/makeAnnotsXml.py and ManageQuickload/quickload/S_lycopersicum_Jun_2022/annots.xml.
Moving to DONE.
I noticed that coverage graphs for this new dataset, which is strand-specific and paired-end, look a bit different, with different patterns of peaks and valleys, compared to earlier data from Genewiz where the data were paired-end and NOT strand-specific. Weird. I don't know why this occurred.
For example, see:
GenomeBrowserImages/TimeCourseVsOlderData-CoverageGraphProfileDifference.png
Creating new ticket to investigate.
Next steps: Find or make a csv sample sheet and change strandedness to 'reverse' to run nextflow.
sample.csv