Details
-
Type:
Epic
-
Status: To-Do (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Unresolved
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Epic Name:Process and deploy Palanivelu Lab data
-
Story Points:4
-
Sprint:Spring 3 2023 Feb 1, Spring 8 2023 Apr 24, Spring 9 2023 May 1
Description
For this task,
- process new and old experimental data sets from the Ravi Palanivelu lab
- confer with Palanivelu lab personnel to understand and document the samples
- track code and key data files using this repository: https://bitbucket.org/hotpollen/pistil-rna-seq/src/main/
About the data:
As of summer 2023, there are now three collections of sequencing data that we got from the Ravi Palanivelu lab. These collections correspond to "batches" of RNA samples that were sent to GeneWiz/Azenta for library synthesis and subsequent sequencing.
For two of these batches, the RP lab created the biological material for samples, extracted RNA, and then sent the RNA boxes to Azenta (formerly GeneWiz), the sequencing company. For one of these batches, the so-called "library synthesis pilot," the RP lab synthesized the libraries themselves and then sent the libraries to Azenta.
Once the sequencing data are complete, the company sent links to an ftp site containing the data files to the RP lab, who downloaded them or asked us to download them. We then obtained the sequence data and deployed them to the Charlotte HPC file system for the next steps - data processing, in which we generate files for visualization in IGB and, also, "counts" files for statistical analysis libraries developed for RNA-Seq data.
The three data collections are:
1) 2021 Kelsie Pryze's unpollinated pistil heat stress experiment, azenta id: 30-681594536
These data are from an experiment done by Kelse Pryze in which she tested the effects of heat stress on un-pollinated tomato pistils dissected from emasculated flowers from four tomato varieties: Heinz, Malintka, Tamaulipas, and Nagcarlang. All sample types have three replicates per sample type represented in the sequencing data, except for Tamaulipas, which has two. KP provided a detailed description of exactly how the samples were generated. The biological material were created in 2021, in the summer and early spring.
The data files also included three data files from a different experiment investigating the transcriptome of dissected, unpollinated ovary tissue. We processed these data alongside the fastq files from the un-pollinated pistils because they were all sequenced at the same time, in the same lot of RNAs sent to the sequencing provider. However, for visualization, we will probably want to present them in ways that will make it super clear that the biological material were created separately from the un-pollinated pistils.
Rob Reid downloaded the data onto the UNCC cluster and saved it here: /projects/tomato_genome/rnaseq/ravi-2022-fullrun/30-681594536.
We then began processing these data in 2023, using the high performance computing cluster at UNC Charlotte. You can identify these samples on our file system by looking for their Azenta identifier - 30-681594536. Also, in our various pipelines and Jira records, we have been referring to these data by the date we got them from the RP Lab: the "Ravi 2022" dataset.
KP provided documentation describing these samples. We will place these files in a "Documentation" folder in the git repository. However, as you will see from the documentation in the repository, the samples themselves were generated during the summer and early spring of 2021.
When we deploy these data to the genome browser for visualization, we will probably use a study name that describes the data and makes it easy for RP Lab personnel and others to recognize them in the browser or other settings.
2) self-pollinated stigma+style heat stress experiment, Azenta id 30-804059537
We have been referring to this experimental data set as "KP-2023", referring to the experimenter Kelsie Pryze and the date when we obtained the experimental data sequence files.
The original sequence files are downloaded to this location on the UNC Charlotte cluster computing system: /projects/tomato_genome/rnaseq/30-804059537-kelsie
This experiment included sample types testing two temperature conditions, three treatment durations, four varieties, and one tissue type. These were:
- temperature conditions: 37 degrees C (heat stress) and 25 degrees C (control)
- treatment durations: 0 hours (no heat stress applied), 3 hours, and 8 hours
- four varieties: Heinz, Malintka, Nagcarlang, Tamaulipas
- tissue type: dissected stigma and style tissue from self-pollinated flowers
There were three replicates per sample type. The zero-hour samples however included three 25 degrees C samples and no 37 degrees C samples.
Number of samples: (2 conditions * 4 varieties * 2 treatment durations * 3 replications) + (1 condition * 4 * 1 treatment duration (0 hours) * 3 replications ) = 60 samples
3) self-pollinated and unpollinated Tamaulipas library preparation pilot experiment, Azenta id 30-605730043
This experiment performed by Kelsie Pryze involved creating libraries for sequencing using RNAs from pollinated and upollinated samples from Tamaulipas plants.
The data from this experiment are stored on the UNC Charlotte cluster in: /projects/tomato_genome/rnaseq/ravi-tamaulipas
Rob downloaded these data from the sequencing provider on or around December 15, 2021. (This is the date that Rob created a Google Doc describing the files available from the sequencing provider's data transfer ftp site.)
Note: We need to confirm if that the sequences obtained from the unpollinated pistils were from the same experiment as (1) above. If yes, which "replicate" were they? This will influence how we label the data in IGB.
To-do for each experimental data set:
- Run nf-core/rnaseq pipeline with both the SL5/2022 and SL4/2019 target genome assemblies using "reverse" strandedness parameter.
- Check the multi-qc report. Re-run the processing as necessary.
- Rename BAM files to not included "sorted" in the name.
- Create scaled coverage graphs.
- Create junction files.
- Migrate data to an on-line location for IGB visualization.
- Create annots.xml metadata file with visualization parameters for each dataset; add the data collection to the makeAnnotsXml.py script
- Add the "counts" data files to the repository for statistical analysis
- Add documentation for each sequence collection to the git repository
- Perform data checking to catch any record-keeping errors that may have occurred
Attached:
- Azenta (sequencing provider) data report for KP 2023 data, with numbers of sequences produced
- Quote from Azenta indicating strand-specific RNA-Seq, 2x150 bp paired end sequencing
Contact:
- Kelsey Pryze - kelseypryze@email.arizona.edu
Attachments
Issue Links
Activity
To use samtools to view the aligned and unaligned.
READS MAPPED:
module load samtools
samtools view -c -F 4 nagcarlang-sorted.bam
For Unmapped:
samtools view -c -f 4 nagcarlang-sorted.bam
Let's calculate coverage:
samtools depth nagcarlang-sorted.bam | awk '
END
{ print "Average = ",sum/NR}'
Put these lines into a slurm script. Should run very quickly.
Created pull request to add src folder and perl validation file to bitbucket:
https://bitbucket.org/hotpollen/pistil-rna-seq/pull-requests/1
[~aloraine]
Created script to use samtools to view the aligned and unaligned:
#!/bin/bash #SBATCH --job-name=samtools_view #job name after submission #SBATCH -p Orion #partition being used #SBATCH -N 1 #number of nodes to use #SBATCH --ntasks-per-node=8 #max number of tasks per node #SBATCH --mem=900gb #memory required per node #SBATCH -t 14-00:00 #time (D-HH:MM) #SBATCH -o samtools_view.%j.out #standard output file #SBATCH --mail-type=END,FAIL #Notifications for job complete/failure #SBATCH --mail-user=mdavi258@uncc.edu #Send to user email #SBATCH --array=1-63 file=$(sed -n -e "${SLURM_ARRAY_TASK_ID}p" /nobackup/tomato_genome/30-804059537-KP/kp_runlist.txt) module load samtools echo "Mapped:" ${file} samtools view -c -F 4 ${file}.bam echo "Unmapped:" ${file} samtools view -c -f 4 ${file}.bam echo "Calculate Coverage" samtools depth ${file}.bam | awk '{sum+=$3} END { print "Average = ",sum/NR}' echo "done" echo "---------------------------------------------------------"
Directory: /nobackup/tomato_genome/30-804059537-KP/results/star_salmon
Combined output files into one:
cat *.out > ./mergedsamtoolsOut.txt
Output File:
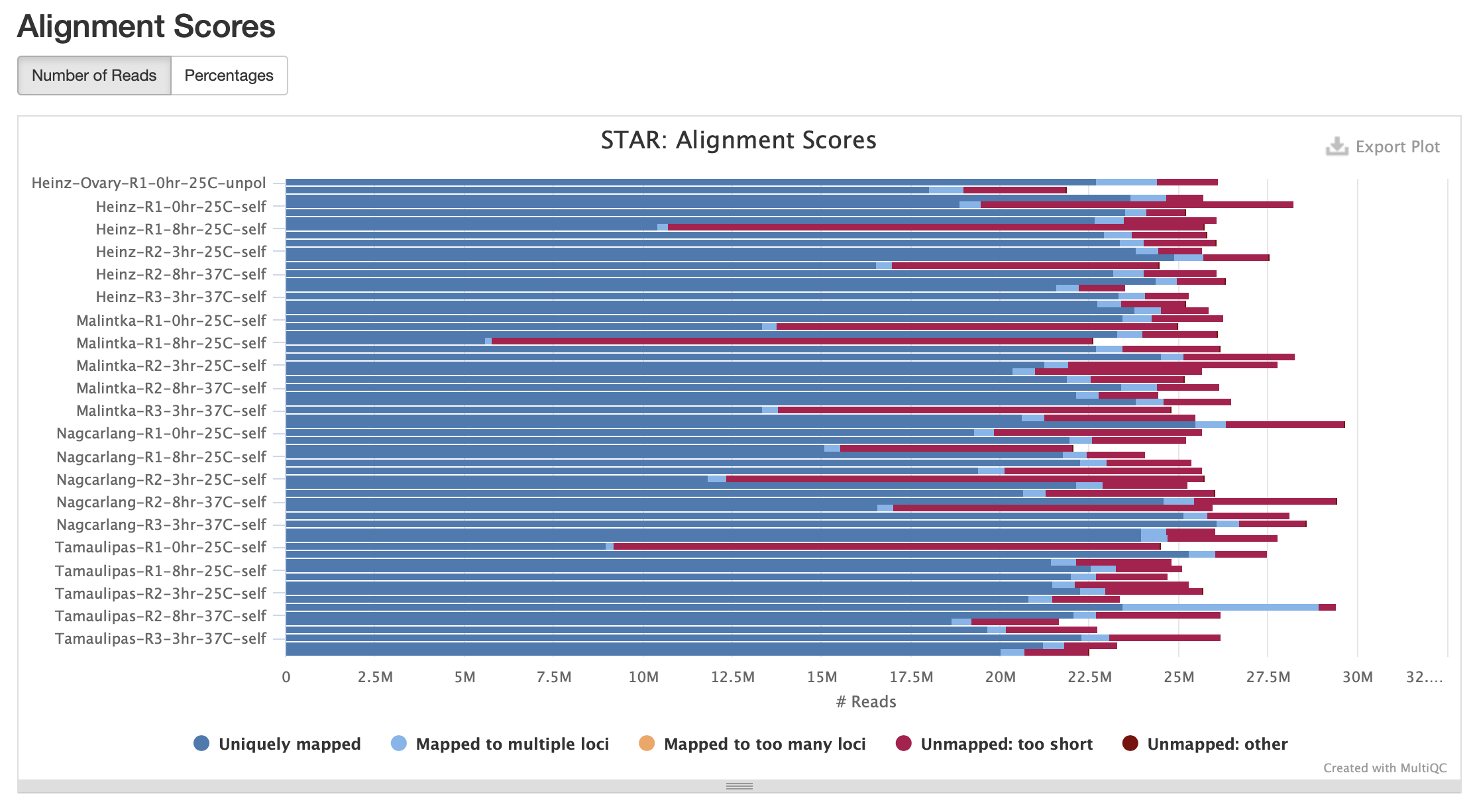
Ann's comments:
Based on output above:
- The bam files do not contain any unmapped reads, only mapped reads
- The samtools "depth" command computes the number of alignments per base pair position - see http://www.htslib.org/doc/samtools-depth.html
- For transcriptome data, the "depth" command does not make a lot of sense because the depth of read alignments at any given position depends on whether or not that position is inside an exon, and also on the level of expression of that exon
- I don't know what "NR" means and where this is coming from in the "sum/NR" statement at the end of the script
Conclusion: This output of this script script does not explain the QC result.
We do not know why some of the samples did not perform well. Let's proceed with the pipeline and visualize the data in a genome browser as this visualization step may reveal more information about the problematic samples.
I am including the Google Links that Kelsie provided here for documentation purposes.
Tomato Pistil Tissue Collection Protocol
https://docs.google.com/document/d/1g8GJBEzxUC-QjfMXk0Eq5mT8e31bGirjXYM3-Sv4u7Q/edit?usp=sharing
Experimental Design for Solavar
https://docs.google.com/document/d/1BXVq-0oop3Ch3Qzbr2nkhQczG1cZH2yBqQRMmBXKGyo/edit?usp=sharing
Sequenced Samples
https://docs.google.com/spreadsheets/d/1WwPzifPzbACmgS3uR_V92cYIGN3qS1yWPGHBu7DY_-I/edit?usp=sharing
Pipeline successfully ran:
Unable to render embedded object: File (Screen Shot 2023-02-09 at 9.40.46 AM.png) not found.
Directory: /nobackup/tomato_genome/30-804059537-KP
Comment: There are no errors in the report but the number of sequences mapped is pretty low. Might need to look into that! Double check sample sheet I made maybe or the wrong reference genome was used to map data.
Unable to render embedded object: File (Screen Shot 2023-02-09 at 10.28.15 AM.png) not found.
Link to interpret report: https://nf-co.re/eager/2.2.2/output#multiqc-report
Next steps: