Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:2
-
Epic Link:
-
Sprint:Fall 1 - Sep 5, Summer 8 2023 Aug 21, Fall 2 2023 Sep 17, Fall 3 2023 Oct 2
Description
My apologies if this has already been done and I lost track of the data somehow!
For this task, we need to align and deploy data from Azenta run 30-804059537 onto the SL4 / S_lycopersicum_Sep_2019 genome, using the S_lycopersicum_Sep_2019 assembly and accompanying gene annotations.
To-do:
- run the nextflow pipeline as per usual. Please use location /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/S_lycopersicum_Sep_2019 on the UNCC cluster
- create a branch in the code repo as usual
- commit to the branch the following files:
30-804059537_SL4_multiqc_report.html (renamed from multiqc_report.html)
30-804059537_SL4_salmon.merged.gene_counts.tsv (renamed from salmon.merged.gene_counts.tsv)
- create coverage graphs and junction files, as per usual
- move just the coverage graphs, bam files, and junction files to this location: /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/for_quickload/S_lycopersicum_Sep_2019/30-804059537 (We'll use this as the source directory for an rsync command that will copy these data over the data.bioviz.org host for visualization in IGB)
Attachments
Issue Links
- relates to
-
-
- Closed
-
Activity
Launch renameBams.sh script:
./renameBams.sh
Launch Scaled Coverage graphs script:
./sbatch-doIt.sh .bam bamCoverage.sh >jobs.out 2>jobs.err
Launch Junction files script:
./sbatch-doIt.sh .bam find_junctions.sh >jobs.out 2>jobs.err
Reviewer:
Check that files have reasonable sizes (no "zero" size files, for example)
Check that every "FJ.bed.gz" file has a corresponding "FJ.bed.gz.tbi" index file
Check that every bam file has a corresponding "FJ.bed.gz" file
Check that every bam file has a corresponding "scaled.bedgraph.gz" file
Check that every "scaled.bedgraph.gz" has a corresponding "scaled.bedgraph.gz.tbi"
Reviewer: [~RobertReid]
Next Steps after review:
- move just the coverage graphs, bam files, and junction files to this location: /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/for_quickload/S_lycopersicum_Sep_2019/30-804059537
- Make sure permissions are granted for copying
Branch: https://bitbucket.org/mdavis4290/molly-pistil-rna-seq/branch/IGBF-3420
Includes 30-804059537_SL4_multiqc_report.html & 30-804059537_SL4_salmon.merged.gene_counts.tsv
Reviewer: [~aloraine]
Checking out files in :
/projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/S_lycopersicum_Sep_2019/results/star_salmon
.err files
All files here are 0 in size.
Except one!
rw-rr- 1 mdavi258 tomato_genome 370 Aug 28 13:56 Malintka-R1-0hr-25C-self.err
The error is
Exception in thread "main" java.lang.RuntimeException: Postion is too high (more than 64792705)
at org.biojava.nbio.genome.parsers.twobit.TwoBitParser.setCurrentSequencePosition(TwoBitParser.java:191)
at org.biojava.nbio.genome.parsers.twobit.TwoBitParser.loadFragment(TwoBitParser.java:332)
at org.lorainelab.findjunctions.FindJunctions.main(FindJunctions.java:249)
Malintka-R1-0hr-25C-self.err lines 1-4/4 (END)
Bam and bai
63 bam files
63 bai files
rw-r---- 1 mdavi258 tomato_genome 590M Aug 28 11:58 Malintka-R1-8hr-25C-self.bam
Sue enough this file is 590M while the rest range from 950M to 3GB. Broken!
Moving this back to "to-do" as the first level review found an issue. MD will look into it, as per her comment during today's morning meeting. Please feel free to move this somewhere different as you see fit, of course.
attn: [~molly]
Nextflow Pipeline ran successfully with SL4 genome
Directory: /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/S_lycopersicum_Sep_2019
MultiQC report notes: No errors or warnings were present in the report. The output file is named '30-804059537_SL4_multiqc_report.html'.
Next steps:
Commit CSV and multiqc report to Pistil repo on bitbucket
Change sorted bam names
Create junction files
Create Coverage graphs
Launch renameBams.sh script:
./renameBams.sh
Launch Scaled Coverage graphs script:
./sbatch-doIt.sh .bam bamCoverage.sh >jobs.out 2>jobs.err
Launch Junction files script:
./sbatch-doIt.sh .bam find_junctions.sh >jobs.out 2>jobs.err
Directory: /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/S_lycopersicum_Sep_2019/results/star_salmon
Still running into same error from before when running find_junctions.sh script: Malintka-R1-0hr-25C-self.err
Exception in thread "main" java.lang.RuntimeException: Postion is too high (more than 64792705)
at org.biojava.nbio.genome.parsers.twobit.TwoBitParser.setCurrentSequencePosition(TwoBitParser.java:191)
at org.biojava.nbio.genome.parsers.twobit.TwoBitParser.loadFragment(TwoBitParser.java:332)
at org.lorainelab.findjunctions.FindJunctions.main(FindJunctions.java:249)
Next Step: Ask Nowlan Freese for help with java jar file (find-junctions-1.0.0-jar-with-dependencies.jar)(S_lycopersicum_Sep_2019.2bit).
Meeting notes:
Issue: Looked at the bam file which looks fine but there are reads that expand upon the chromosome. They have been soft clipped but find_junctions isn't taking that into consideration and won't process the file entirely to create the .FJ.bed.gz file.
Extra notes: There is a read 64792,798 of the actual chromosome that is past the end of the SL4 genome. The reader should have clipped the read but didn't do this and now is not fitting correctly at the ends. The alignment didn't clip the ends to match the SL4 genome correctly just for this one sample Malintka-R1-0hr-25C-self. Junctions confused because what was soft clipped did pass but find_junctions for this bam file is unusable. Find_junctions isn't obeying that the files have been soft clipped. Aligner should hard-clip but instead is soft-clipping.
Code used:
SL4.0ch10:64782127-64791136 > samtoolsOutput.bam java -jar find-junctions-1.0.0-jar-with-dependencies.jar -u -f 5 -b S_lycopersicum_Sep_2019.2bit -o myShortFileoutput.bed samtoolsOutput.bamsamtools view -b Malintka-R1-0hr-25C-self.bam
Possible solution: Change code to find_junctions to allow soft-clipping if over the distance required. But this is for only one file so would it be worth changing the code?
Could also review the multiqc report to maybe discover clipping issue: Per Base Sequence Content has failed and usually means there are contaminates in the library. Aligners should be able to deal with bases that don't align and soft-clip them. But in this case for this one sample it did not do this correctly?
Ann comment: Identify the aligned region and visualize it in IGB. Document and capture the image for presentations.
From meeting: Re-run / double-check on Malintka-R1-8hr-25C-self (not the same sample that messed up the find junctions program.)
New ticket was made for junctions java error: IGBF-3434
Next step for this ticket: Review Malintka-R1-8hr-25C-self.bam file size issue and see if the problem is repetitive after nextflow rerun
Malintka-R1-8hr-25C-self.bam
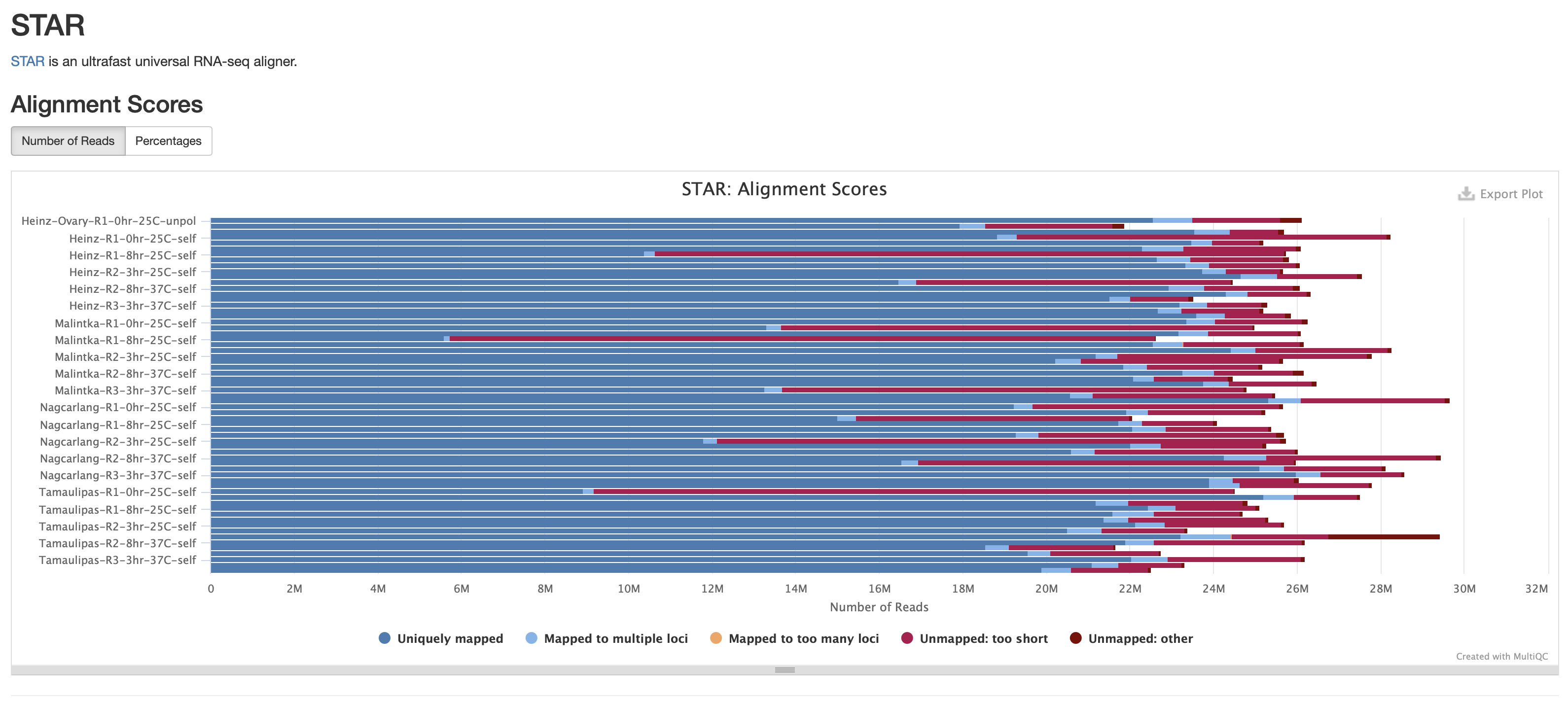
- The files does seem to be very small (618061348M) compared to other bam files on the cluster. I checked the multiQC report and this sample does have the worst alignment score from unmapped reads due to them being too short. This means that this isn't a pipeline issue but more of a sample issue.
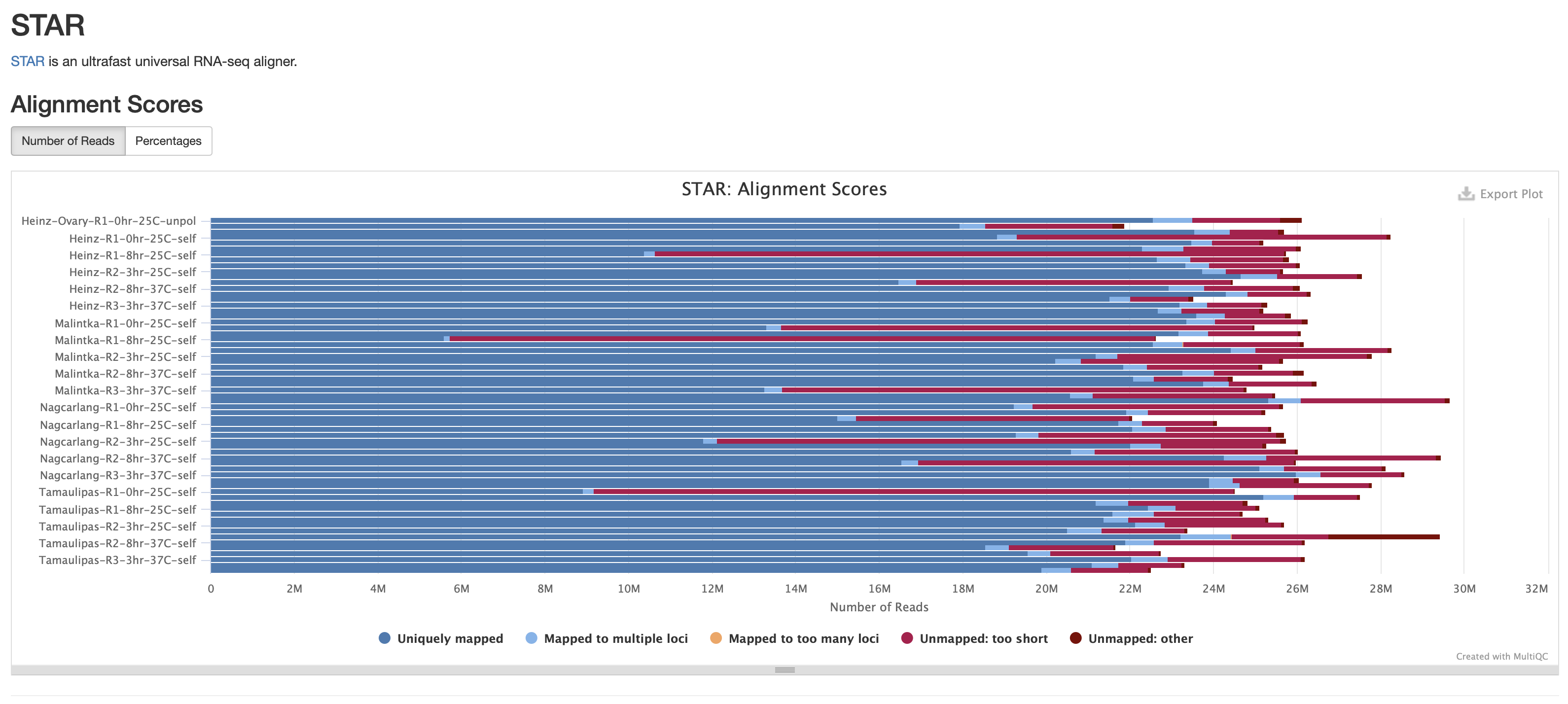
Next Step: Please review my conclusions and move ticket to done if there are no issues.
I took a peak at the number of reads that align for each expt.
Total reads that align:
Total Read Count
Heinz-Ovary-R1-0hr-25C-unpol.bam 50979464
Heinz-Ovary-R2-0hr-25C-unpol.bam 39862409
Heinz-Ovary-R3-0hr-25C-unpol.bam 52051210
Heinz-R1-0hr-25C-self.bam 40438938
Heinz-R1-3hr-25C-self.bam 49881450
Heinz-R1-3hr-37C-self.bam 51660872
Heinz-R1-8hr-25C-self.bam 22196798
Heinz-R1-8hr-37C-self.bam 50926978
Heinz-R2-0hr-25C-self.bam 49858292
Heinz-R2-3hr-25C-self.bam 50700025
Heinz-R2-3hr-37C-self.bam 54923225
Heinz-R2-8hr-25C-self.bam 35292739
Heinz-R2-8hr-37C-self.bam 51480404
Heinz-R3-0hr-25C-self.bam 51577031
Heinz-R3-3hr-25C-self.bam 45984816
Heinz-R3-3hr-37C-self.bam 50612275
Heinz-R3-8hr-25C-self.bam 48707431
Heinz-R3-8hr-37C-self.bam 51695156
Malintka-R1-0hr-25C-self.bam 50644328
Malintka-R1-3hr-25C-self.bam 28575876
Malintka-R1-3hr-37C-self.bam 50707327
Malintka-R1-8hr-25C-self.bam 11997289
Malintka-R1-8hr-37C-self.bam 49495351
Malintka-R2-0hr-25C-self.bam 52432853
Malintka-R2-3hr-25C-self.bam 45473568
Malintka-R2-3hr-37C-self.bam 44168728
Malintka-R2-8hr-25C-self.bam 46900467
Malintka-R2-8hr-37C-self.bam 51370963
Malintka-R3-0hr-25C-self.bam 47086317
Malintka-R3-3hr-25C-self.bam 51085380
Malintka-R3-3hr-37C-self.bam 29231411
Malintka-R3-8hr-25C-self.bam 44275394
Malintka-R3-8hr-37C-self.bam 55644007
Nagcarlang-R1-0hr-25C-self.bam 40968404
Nagcarlang-R1-3hr-25C-self.bam 46600257
Nagcarlang-R1-3hr-37C-self.bam 32781471
Nagcarlang-R1-8hr-25C-self.bam 46430696
Nagcarlang-R1-8hr-37C-self.bam 49122203
Nagcarlang-R2-0hr-25C-self.bam 41919616
Nagcarlang-R2-3hr-25C-self.bam 25538537
Nagcarlang-R2-3hr-37C-self.bam 48518069
Nagcarlang-R2-8hr-25C-self.bam 44389303
Nagcarlang-R2-8hr-37C-self.bam 55567552
Nagcarlang-R3-0hr-25C-self.bam 35216477
Nagcarlang-R3-3hr-25C-self.bam 53420765
Nagcarlang-R3-3hr-37C-self.bam 55258962
Nagcarlang-R3-8hr-25C-self.bam 50940477
Nagcarlang-R3-8hr-37C-self.bam 51768199
Tamaulipas-R1-0hr-25C-self.bam 19287669
Tamaulipas-R1-3hr-25C-self.bam 54518901
Tamaulipas-R1-3hr-37C-self.bam 47761377
Tamaulipas-R1-8hr-25C-self.bam 48604790
Tamaulipas-R1-8hr-37C-self.bam 50484499
Tamaulipas-R2-0hr-25C-self.bam 46447680
Tamaulipas-R2-3hr-25C-self.bam 48552903
Tamaulipas-R2-3hr-37C-self.bam 46727628
Tamaulipas-R2-8hr-25C-self.bam 56810522
Tamaulipas-R2-8hr-37C-self.bam 48005344
Tamaulipas-R3-0hr-25C-self.bam 40520115
Tamaulipas-R3-3hr-25C-self.bam 42195143
Tamaulipas-R3-3hr-37C-self.bam 49785458
Tamaulipas-R3-8hr-25C-self.bam 45882212
Tamaulipas-R3-8hr-37C-self.bam 44242182
Script that does this is here:
/projects/tomato_genome/scripts/rob/summarizeBAMAlignments.slurm
Compared to the average number of reads aligned, the 3 expts with much lower total reads are:
name num aligned Percent of average
Tamaulipas-R1-0hr-25C-self.bam 19287669 -57.8
Malintka-R1-8hr-25C-self.bam 11997289 -73.7
Heinz-R1-8hr-25C-self.bam 22196798 -51.4
Going to explore next if this pattern is observed throughout NEXTFLOW and post processing.
Bam checking shows the BAMs seem intact. The temporary steps in NEXTFLOW are lost as part of the post cleanup so we can't peak at what happens mid process.
I left a copy of the slurm script in:
/projects/tomato_genome/scripts/rob/summarizeBAMAlignments.slurm
So Bam files seem fine at this stage.
Had to rebase my branch:
git checkout IGBF-3420 git pull --rebase upstream main git push --force origin IGBF-3420
Branch: https://bitbucket.org/mdavis4290/molly-pistil-rna-seq/branch/IGBF-3420
Pull Request: https://bitbucket.org/hotpollen/pistil-rna-seq/pull-requests/9
Note: Files should include SL4 Multiqc report and counts.tsv file
Just moved all coverage graphs, bam files, and junction files to this location: /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/for_quickload/S_lycopersicum_Sep_2019/30-804059537
Permission note:
'- rw-rw-r- -' (first rw references owner can read and write, next rw references group can read and write, next r references everyone)
Suggestions for testing:
- View the multi_qc report, compare to SL5 counterpart
- Check that the gene counts file has the expected number of lines (same as equivalent files from other SL4 data processing)
Testing:
- Comparing SL4 and SL5 multiqc report: Both reports are pretty similar. of course mapping differs between each genome but overall there are no major differences.
- The counts.tsv file has the correct amount of rows for genes. This was compared with muday counts.tsv SL4 output data.
- One note is that the SL5 counts.tsv file is not present in the repo.
Branch: https://bitbucket.org/mdavis4290/molly-pistil-rna-seq/branch/IGBF-3420b
Adds file: ExternalData/30-804059537-SL5-salmon.merged.gene_counts.tsv
Beginning with review of all things in branch above.
Took a quick glance at the .tsv salmon file in the recent branch and it appears intact and correct, i.e., # of rows expected and # of columns.
Robust counts! Lots of things going on expression wise. This might be a very interesting dataset!
Moving on to the cluster ( /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/for_quickload/S_lycopersicum_Sep_2019/30-804059537) to check those bits.
In /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/for_quickload/S_lycopersicum_Sep_2019/30-804059537
As expected:
63 bams
63 bais
126 .gz files (63 bedgraphs, 63 beds)
126 .tbi files to go with the bed files.
The BAM files have a wide range of sizes which at this point we are thinking is just how those sequences are.
Taking a quick peak at the raw files, /projects/tomato_genome/rnaseq/30-804059537-kelsie/00_fastq$
The size of these fastq files are not as wide ranging as the BAM files are.
Makes me think that either the BAM alignment had issues on some samples (less likely, but we will find out later when we rerun these as SRA's) or something in the sample prep went awry and there is contamination. To test this we could run STAR, and get all the reads that do not align. I think NEXTFLOW is deleting the unaligned reads.
If we get our hands on unaligned reads we can blast against the NR DB and see if there is other things that got sequenced, like for example the technician!
I'll review more soon.
For this:
https://bitbucket.org/mdavis4290/molly-pistil-rna-seq/branch/IGBF-3420b
Is all that is needed is to check out the .tsv file?
I'll assume so and bounce this ticket back to Molly!
Pull Request: https://bitbucket.org/hotpollen/pistil-rna-seq/pull-requests/10
Note: Adds 30-804059537-SL5-salmon.merged.gene_counts.tsv to external folder with SL4 tsv file so comparisons can be made.
Merged the PR but then changed name from 30-804059537-SL5-salmon.merged.gene_counts.tsv to 30-804059537-SL5_salmon.merged.gene_counts.tsv to match convention established for other such files throughout the project.
Directory: /projects/tomato_genome/fnb/dataprocessing/30-804059537-KP/S_lycopersicum_Sep_2019