Details
-
Type:
New Feature
-
Status: Open (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Unresolved
-
Labels:None
-
Epic Link:
Description
Background: In epigenetic studies, researchers often look for methylation. Methylation is when a methyl group has been added to a cytosine. In order to determine if methylation is present, a process called bisulfite conversion is used. Bisulfite conversion converts all non-methylated cytosines to uracils - which are then read as thymines during sequencing.
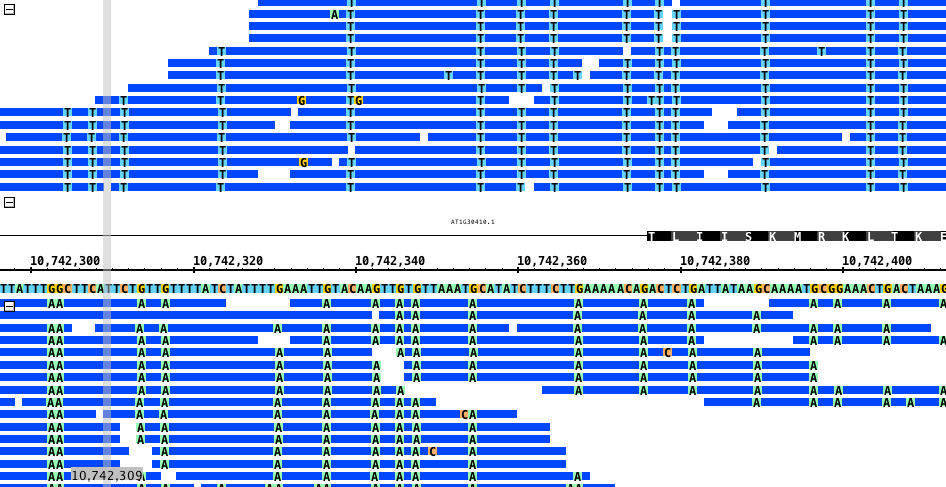
Current view in IGB: Bisulfite converted reads appear as mismatches in IGB due to the majority of cytosines not being methylated. These show up as thymines on the positive strand (in place of cytosine), and adenines on the negative strand (in place of guanines). (See image 1 and 6)
New implementation in IGB: A user would right click on the track of interest and select Show Methylation, which would have an arrowed menu that would allow the user to select multiple instances of methylation (CG, CHH, etc.). This would differentially color cytosines based on their methylation status. This would allow IGB users to quickly scan through their aligned read file looking for trends in methylation. The user could then turn off the Show Methylation mode by unchecking the menu.
Additional Note:
Programs that output bisulfite sequenced alignment files (bam or sam) usually include additional information within the file specifying the methylation status for each read, at least in terms of instances of methylation for CG, CHG, and CHH.
Output from the bisulfite sequence aligner Bismark has this format:
(1) QNAME (seq-ID)
(2) FLAG (this flag tries to take the strand a bisulfite read originated from into account (this is different from ordinary DNA alignment flags!))
(3) RNAME (chromosome)
(4) POS (start position)
(5) MAPQ (only calculated for Bowtie 2, always 255 for Bowtie)
(6) CIGAR
(7) RNEXT
(8) PNEXT
(9) TLEN
(10) SEQ
(11) QUAL (Phred33 scale)
(12) NM-tag (edit distance to the reference)
(13) MD-tag (base-by-base mismatches to the reference)
(14) XM-tag (methylation call string)
(15) XR-tag (read conversion state for the alignment)
(16) XG-tag (genome conversion state for the alignment)
Note the XM, XR, and XG tags. For the XM tag, the individual "." equate to a base position in the read, whereas x, z, or h equate to methylation calls for the following instances:
z unmethylated C in CpG context
Z methylated C in CpG context
x unmethylated C in CHG context
X methylated C in CHG context
h unmethylated C in CHH context
H methylated C in CHH context
I've also included an example .sam file of the aligned output from Bismark.