Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:2
-
Epic Link:
-
Sprint:Fall 7 2022 Nov 21, Fall 8 2022 Dec 5, Spring 1 2023 Dec 26, Spring 2 2023 Jan 16, Spring 3 2023 Feb 1, Spring 4 2023 Feb 21
Attachments
Issue Links
Activity
Updated Multiqc report:
Final_SRP268884_multiqc_report.html![]()
Updated Bitbucket CSV file:
https://bitbucket.org/hotpollen/splicing-analysis/pull-requests/9
Updated Scaled Coverage Graphs:
/nobackup/tomato_genome/alt_splicing/for_igbquickload/coverage_graphs_2/coverage_graphs_SRP268884
Processing parameters additionally include "tomato.config" file:
params {
modules {
'star_align' {
args = '--alignIntronMax 13000 --quantMode TranscriptomeSAM --twopassMode Basic --outSAMtype BAM
Unsorted --readFilesCommand zcat --runRNGseed 0 --outFilterMultimapNmax 20 --alignSJDBoverhangMin 1 --outSAMattributes N
H HI AS NM MD --quantTranscriptomeBan Singleend'
}
'hisat2_align' {
args = "--max-intronlen 13000 --met-stderr --new-summary --dta"
}
}
}
Adding multiqc files to repository hotpollen/splicing-analysis.git.
Copied files to renci host:
- SRP100604 coverage graphs
- SRP100604 alignment files
- SRP100604 junction files
- SRP268884 coverage graphs
- SRP268884 alignment files
- SRP268884 junction files
Example invocation:
scp -J aloraine@hop.renci.org -r SRP268884_bam aloraine@lorainelab-quickload.scidas.org:/projects/igbquickload/lorainelab/www/main/htdocs/hotpollen/S_lycopersicum_Jun_2022/SRP268884/.
All files are made, transferred to RENCI for hosting. Moving to DONE.
SRP100604 and SRP268884 have been uploaded and fastq files created.
SRA links:
https://www.ncbi.nlm.nih.gov/sra?term=SRP100604
https://www.ncbi.nlm.nih.gov/sra?term=SRP268884
Code Example
prefetch.slurm
#! /bin/bash #SBATCH --job-name=prefetch_SRR #SBATCH --partition=Orion #SBATCH --nodes=1 #SBATCH --ntasks-per-node=1 #SBATCH --mem=4gb #SBATCH --output=%x_%j.out #SBATCH --time=24:00:00 cd /nobackup/tomato_genome/alt_splicing/SRP100604 module load sra-tools/2.11.0 vdb-config --interactive files=( SRR5279858 SRR5279875 SRR5279883 SRR5280323 SRR5280370 SRR5280382 SRR5280383 SRR5280392 SRR5282476 SRR5282478 SRR5282480 SRR5282481 ) for f in "${files[@]}"; do echo $f; prefetch $f; donefasterdump.slurm
Comments on results
Directory: /nobackup/tomato_genome/alt_splicing/SRP100604
SRP100604: There were some SRR files that were not double stranded but were single stranded so it could not make _1.fastq and _2.fastq files.
List of those SRR's-
SRR5282476
SRR5282478
SRR5282480
SRR5282481
Directory: /nobackup/tomato_genome/alt_splicing/SRP268884
SRP268884: Produces all double stranded _1.fastq and _2.fastq files.
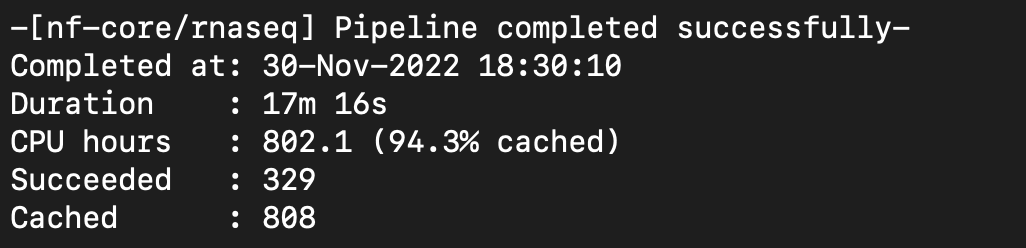
Next Step: Run Nextflow rnaseq/nf-core pipeline on SRP268884.
Question: Should we still use SRP100604 if it contains single stranded SRR files or just use the double stranded files that it contained?
[~aloraine]
aloraine's answer to the above query: Go ahead and use all the available data in SRP100604. I believe that nextflow is able to handle this complication intelligently. I think you can omit the "second" file name in the "samples" file for single end runs. (Please note that "single strand" is not the same thing as "single end" - make sure that we are talking about the same thing before proceeding.)