Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:2
-
Epic Link:
-
Sprint:Fall 7 2022 Nov 21, Fall 8 2022 Dec 5, Spring 1 2023 Dec 26, Spring 2 2023 Jan 16, Spring 3 2023 Feb 1, Spring 4 2023 Feb 21
Attachments
Issue Links
Activity
Nextflow Update:
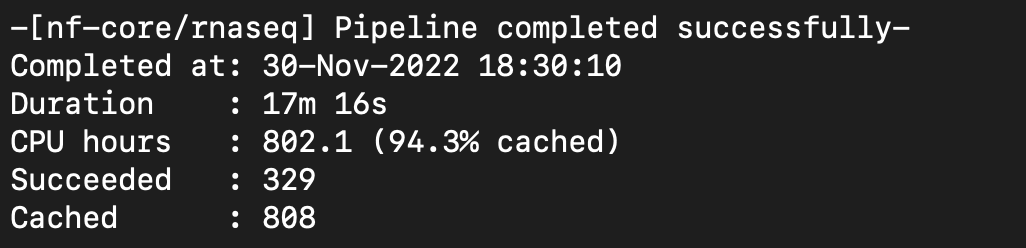
Pipeline ran successfully with SRP268884.
Notes: Could not find whether data was stranded or unstranded on csv file. Went ahead and just put stranded on csv file. To discover that you can run an alignment first and view files in IGB. Stranded = no mismatch of stranded.
Nextflow Notes: Took a couple times for nextflow to run. It would randomly stop and throw a random error and I would just run it again without changing anything and it would work.
[~aloraine]
SRP100604 Update:
Directory: /nobackup/tomato_genome/alt_splicing/SRP100604
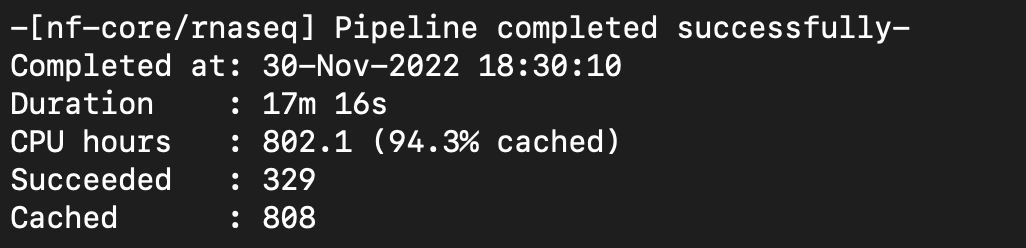
Nextflow ran successfully with SRP100604. I made a csv sample sheet with paired and single end fastq files.
Notes: No issues with Nextflow this time. I am having trouble getting rsync to work with my local machine to view output files to determine whether the projects are stranded or unstranded. Will have to probably contact researchers to get that information if I can't get rsync to work.
Resources/Instruction:
Stranded or unstranded- https://nf-co.re/rnaseq/1.2/output#rseqc
How to make .csv sample sheet for single and paired- https://nf-co.re/rnaseq/usage
Next step: I need to fix names for bam files so only the SRR names are there.
[~aloraine]
Update: I have renamed the bam files and reviewed the results from the nextflow pipeline. I added the sample csv files used for nextflow to bitbucket.
Next Steps: The files need to be reviewed by someone on the team please. The next task is to make description sheets for both samples (IGBF-3238).
[~aloraine]
In reviewing the PR for the sample sheets, I found what looks like a possible problem. Here is the comment I made on the PR regarding this possible problem:
I have a concern about this line: SRR5282476,SRR5282476.fastq.gz,SRR5282476.fastq.gz,unstranded
In this case, the sample run (SRR5282474) was sequenced on only one end, making it a “single end” not “paired end” dataset. In this case, I believe what you need to do with nextflow is simple omit field 3, like so:
SRR5282476,SRR5282476.fastq.gz,,unstranded
I’m concerned about what happens to the “run” if you do not do this. For example, the software may run incorrectly.
Just to be on the safe side, I think it would be a good idea to re-run the nextflow pipeline for this entire set of samples.
Also, another way to assess the situation is to look at the output of the multiqc processing. This might be helpful.
I reviewed the files I submitted to bitbucket and SRP100604 needed to be updated to the new one I manually fixed on the cluster. The results were used with the correct updated file and I believe there is no need for a re-run of nextflow due to the right files being used. I have updated bitbucket with the correct updated file now and resubmitted the pull request.
https://bitbucket.org/hotpollen/splicing-analysis/pull-requests/8
Update: The pull request was merged with the correct csv files.
Updated PR is now merged to branch "main."
Comment: Need to check MultiQC report to check strandedness. Having trouble with scp and rsync so I haven't been able to download from cluster and check myself.
I agree with the previous comment.
Next steps:
- Download multiqc reports
- Rename each to use the SRP number, e.g., SRP268884_multiqc_report.html
- Attach to this ticket
- Review the content
To download, use scp.
Example invocation - on your local:
scp aloraine@hpc.uncc.edu:/nobackup/tomato_genome/alt_splicing/SRP268884/results/multiqc/star_salmon/multiqc_report.html SRP268884_multiqc_report.html
- When do this, you need to authorize via the usual two-factor authentication.
- The above example command renames the downloaded file.
- To download and keep the same file name, replace the second argument with "."
Notes: Downloaded multiqc reports on local machine with code-
scp mdavi258@hpc.uncc.edu:/nobackup/tomato_genome/alt_splicing/SRP268884/results/multiqc/star_salmon/multiqc_report.html SRP268884_multiqc_report.html
[^SRP268884_multiqc_report.html]
SRP100604_multiqc_report.html![]()
Update: I need to rerun SRP268884 and fix csv sample strandedness to 'reverse'. After successfully running I need to review multiqc report and add it to this ticket, update the csv file on bitbucket, remove sorted names, and redo scaled coverage graphs for SRP268884.
Side Note: I would like to improve my skills in reading and understanding multiqc reports.
Updated Multiqc report:
Final_SRP268884_multiqc_report.html![]()
Updated Bitbucket CSV file:
https://bitbucket.org/hotpollen/splicing-analysis/pull-requests/9
Updated Scaled Coverage Graphs:
/nobackup/tomato_genome/alt_splicing/for_igbquickload/coverage_graphs_2/coverage_graphs_SRP268884
Processing parameters additionally include "tomato.config" file:
params {
modules {
'star_align' {
args = '--alignIntronMax 13000 --quantMode TranscriptomeSAM --twopassMode Basic --outSAMtype BAM
Unsorted --readFilesCommand zcat --runRNGseed 0 --outFilterMultimapNmax 20 --alignSJDBoverhangMin 1 --outSAMattributes N
H HI AS NM MD --quantTranscriptomeBan Singleend'
}
'hisat2_align' {
args = "--max-intronlen 13000 --met-stderr --new-summary --dta"
}
}
}
Adding multiqc files to repository hotpollen/splicing-analysis.git.
Copied files to renci host:
- SRP100604 coverage graphs
- SRP100604 alignment files
- SRP100604 junction files
- SRP268884 coverage graphs
- SRP268884 alignment files
- SRP268884 junction files
Example invocation:
scp -J aloraine@hop.renci.org -r SRP268884_bam aloraine@lorainelab-quickload.scidas.org:/projects/igbquickload/lorainelab/www/main/htdocs/hotpollen/S_lycopersicum_Jun_2022/SRP268884/.
All files are made, transferred to RENCI for hosting. Moving to DONE.
SRP100604 and SRP268884 have been uploaded and fastq files created.
SRA links:
https://www.ncbi.nlm.nih.gov/sra?term=SRP100604
https://www.ncbi.nlm.nih.gov/sra?term=SRP268884
Code Example
prefetch.slurm
#! /bin/bash #SBATCH --job-name=prefetch_SRR #SBATCH --partition=Orion #SBATCH --nodes=1 #SBATCH --ntasks-per-node=1 #SBATCH --mem=4gb #SBATCH --output=%x_%j.out #SBATCH --time=24:00:00 cd /nobackup/tomato_genome/alt_splicing/SRP100604 module load sra-tools/2.11.0 vdb-config --interactive files=( SRR5279858 SRR5279875 SRR5279883 SRR5280323 SRR5280370 SRR5280382 SRR5280383 SRR5280392 SRR5282476 SRR5282478 SRR5282480 SRR5282481 ) for f in "${files[@]}"; do echo $f; prefetch $f; donefasterdump.slurm
Comments on results
Directory: /nobackup/tomato_genome/alt_splicing/SRP100604
SRP100604: There were some SRR files that were not double stranded but were single stranded so it could not make _1.fastq and _2.fastq files.
List of those SRR's-
SRR5282476
SRR5282478
SRR5282480
SRR5282481
Directory: /nobackup/tomato_genome/alt_splicing/SRP268884
SRP268884: Produces all double stranded _1.fastq and _2.fastq files.
Next Step: Run Nextflow rnaseq/nf-core pipeline on SRP268884.
Question: Should we still use SRP100604 if it contains single stranded SRR files or just use the double stranded files that it contained?
[~aloraine]
aloraine's answer to the above query: Go ahead and use all the available data in SRP100604. I believe that nextflow is able to handle this complication intelligently. I think you can omit the "second" file name in the "samples" file for single end runs. (Please note that "single strand" is not the same thing as "single end" - make sure that we are talking about the same thing before proceeding.)