Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:0.5
-
Epic Link:
-
Sprint:Spring 2 2023 Jan 16
Description
Re-run nextflow rnaseq pipeline in:
/nobackup/tomato_genome/seedlingPollen/nfcore
Using new samples.csv file with "reverse" in the strandedness field.
Please just edit "samples.csv" in /nobackup/tomato_genome/seedlingPollen/nfcore.
Note about sample names:
MP - mature pollen
Se - seedling
N - nagcarlang
H - heinz
T - tamaulipas
M - malintka
S - stress
C - control
See folder in the google drive with info about the experiment:
- Experiments > Pollen Grain and Seedling RNA-seq
Attachments
Issue Links
- relates to
-
-
- Closed
-
Activity
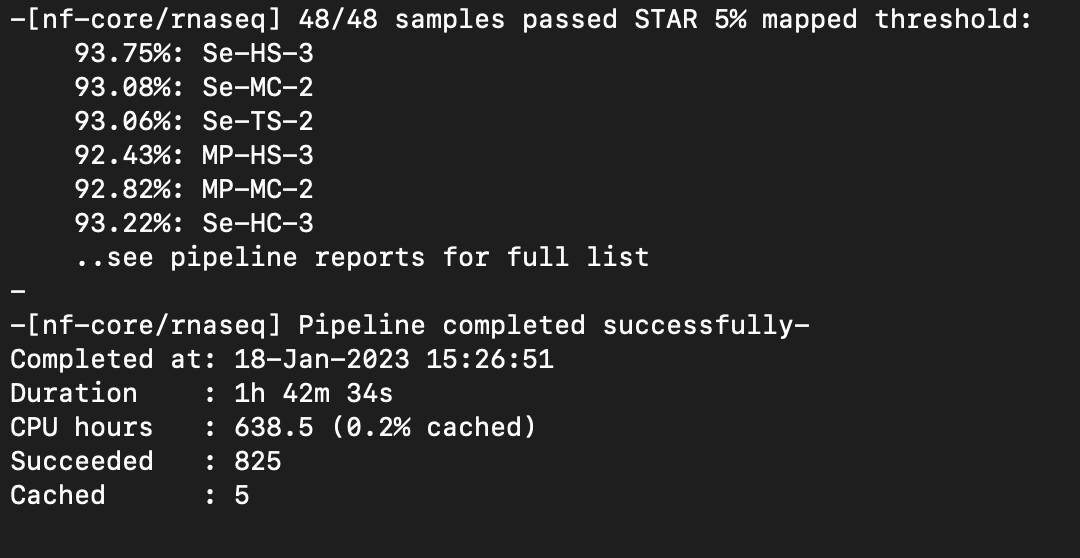
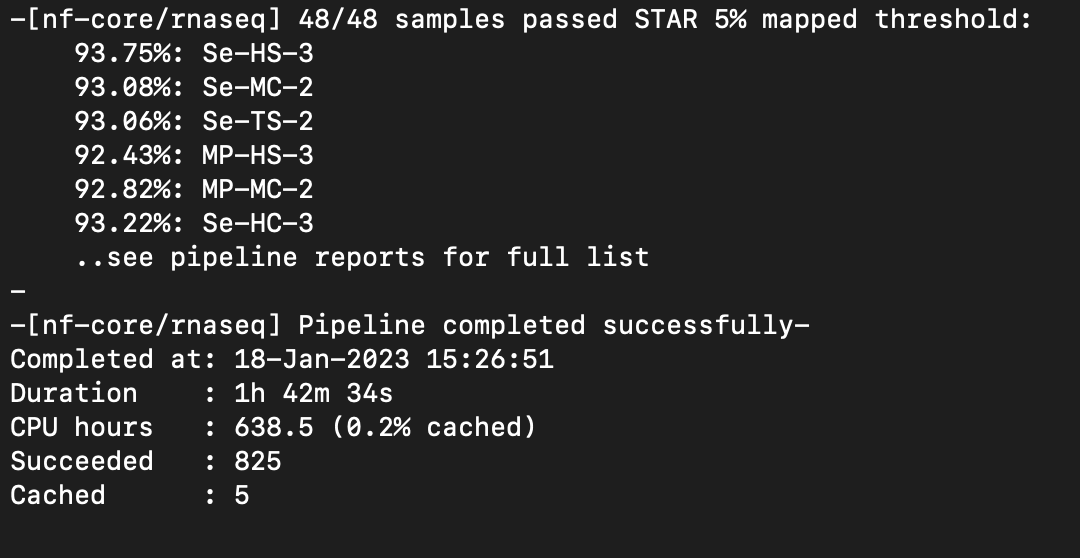
Nextflow Pipeline Ran Successfully!
Directory: /nobackup/tomato_genome/seedlingPollen/nfcore
Next steps: Rename sorted bam files and make scaled coverage graphs.
Scaled coverage graphs have been made and are located:
/nobackup/tomato_genome/seedlingPollen/nfcore/results/star_salmon
Notes: I can move the coverage graphs to their own directory if you would like. Let me know!
Multiqc Report:
scp mdavi258@hpc.uncc.edu:/nobackup/tomato_genome/seedlingPollen/nfcore/results/multiqc/star_salmon/multiqc_report.html nfcore_multiqc_report.html
Notes: Seems there are no issues with strandedness.
Next step: Pipeline, coverage graphs, and Multiqc report need to be reviewed.
[~aloraine]
Ran find junctions pipeline. Copied data to host with:
scp -J aloraine@hop.renci.org -r junctions aloraine@lorainelab-quickload.scidas.org:/projects/igbquickload/lorainelab/www/main/htdocs/hotpollen/S_lycopersicum_Jun_2022/mark-2022-timeseries/.
Added samples.csv file to repository. See: https://bitbucket.org/hotpollen/splicing-analysis/src/main/ExternalData/seedlingPollen-samples.csv
Got description of data set. See attached.
Deployed data to quickload. Did not examine pattern of coverage graphs relative to other data sets.
Added sample sheet to repositor: https://bitbucket.org/hotpollen/splicing-analysis/src/main/ExternalData/seedlingPollen_sample_sheet.xlsx
Update: I fixed the sample sheet so that strandedness is reverse. When running nextflow I come across this error for trimgalore:
Question: are all fastq files double stranded or single stranded? Or is there a naming issue in the sample csv file?
Next step: Instead of using symbolic links I will copy the fastq files to the following directory to see if it works with nextflow. I have had issues with symbolic links before with fastq files and copying the files instead seemed to fix the issue.
Copy From: /projects/tomato_genome/rnaseq/mark-2022-pollengrainSeedling/00_fastq/
Copy To: /nobackup/tomato_genome/seedlingPollen/nfcore
Issue: Do not have permission to copy fastq files.
Fixed Issue: Dr. Robert Reid gave permission so I could copy files over.