Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:3
-
Epic Link:
-
Sprint:Spring 6 2023 Mar 20, Spring 7 2023 Apr 10, Spring 8 2023 Apr 24
Description
This data set from Arabidopsis thaliana contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen.
For this task
- download the data as fastq files from SRA
- align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below)
- for alignment parameters, use original publication (referenced above) and their parameters that they used in their experiment. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species!
- align using same maxIntron parameter reported in the methods section for the paper
- for the above, make a new "config" file
- create coverage graphs
- create junction files
Use this reference genome for alignment:
Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.)
2bitToFa command:
twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa
Attachments
Issue Links
- relates to
-
-
- To-Do
-
Activity
Copied files to /nobackup/tomato_genome/alt_splicing/SRP371294.transfer ( a location where my user has write permission )
Number of files: 36
Number of samples: 6
Size: 15 Gb
Deploying data to:
/projects/igbquickload/lorainelab/www/main/htdocs/rnaseq/A_thaliana_Jun_2009/SRP371294
On RENCI sci-das host.
Note:
This will not be part of the hotpollen Quickload but instead will get put into the "rnaseq" quickload.
Transferring with:
scp -J aloraine@hop.renci.org -r SRP371294.transfer aloraine@lorainelab-quickload.scidas.org:/projects/igbquickload/lorainelab/www/main/htdocs/rnaseq/A_thaliana_Jun_2009/SRP371294/.
Added files to repository:
- SRP371294-multiqc_report.html
- SRP371294-salmon.merged.gene_counts.tsv
- SRP371294.config (copy of /nobackup/tomato_genome/alt_splicing/SRP371294/Arabidopsis.config)
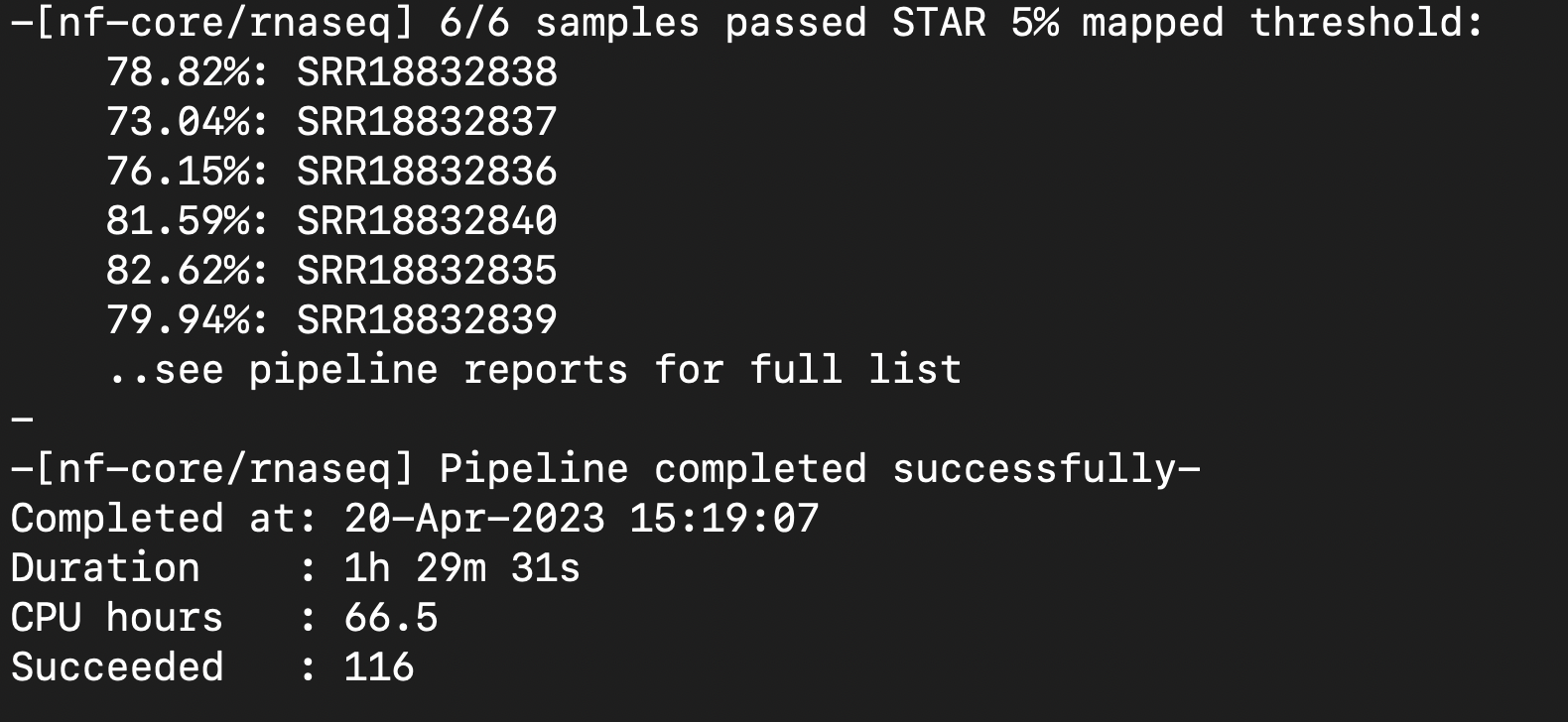
Multiqc file looks fine.
Moving to DONE.
Directory: /nobackup/tomato_genome/alt_splicing/SRP371294
Config parameters from paper that published results(link in description): “–dta –min-intronlen 60 –max-intronlen 6000”
params { modules { 'star_align' { args = '--alignIntronMax 6000 --quantMode TranscriptomeSAM --twopassMode Basic --outSAMtype BAM Unsorted --readF$ } 'hisat2_align' { args = " --max-intronlen 6000 --met-stderr --new-summary --dta" } } }GTF file: https://www.arabidopsis.org/download/index-auto.jsp?dir=%2Fdownload_files%2FGenes%2FAraport11_genome_release
Bed file: Location of bed file?
See: http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/Araport11.bed.gz.
Note that this is a "BED-detail" file and has fields 13 and 14. The nfcore/rnaseq pipeline may not accept this. You may need to modify it to remove fields 13 and 14.
CSV file: Make sure to check mulitqc report to see if strandedness is correct!