Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:3
-
Epic Link:
-
Sprint:Spring 6 2023 Mar 20, Spring 7 2023 Apr 10, Spring 8 2023 Apr 24
Description
This data set from Arabidopsis thaliana contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen.
For this task
- download the data as fastq files from SRA
- align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below)
- for alignment parameters, use original publication (referenced above) and their parameters that they used in their experiment. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species!
- align using same maxIntron parameter reported in the methods section for the paper
- for the above, make a new "config" file
- create coverage graphs
- create junction files
Use this reference genome for alignment:
Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.)
2bitToFa command:
twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa
Attachments
Issue Links
- relates to
-
-
- To-Do
-
Activity
Need approval from [~aloraine] to decide which GTF file to use.
Making GTF file:
- Downloaded Araport11.bed.gz and added to splicing-analysis repository
- Used code from repository genomesource to make a GTF version of Araport.bed.gz:
gunzip -c ~/src/splicing-analysis/ExternalData/Araport11.bed.gz | bed2gtf.py -g 4 > ~/src/splicing-analysis/ExternalData/Araport11.gtf
Repository with bed2gtf.py:
local aloraine$ git remote -v origin git@bitbucket.org:lorainelab/genomesource.git (fetch)
Version of bed2gtf.py used:
5dd6d47 (HEAD -> master, origin/master, origin/HEAD) IGBF-3256 Make conversation file executable 92eb050 IGBF-3256 Support CHESS GFF format 34eac2e Improve GTF to/from BED conversion
Oops! "conversation" should be "conversion"
GTF file is created and added to hotpollen/splicing-analysis as ExternalData/Araport11.gtf.
attn: [~molly]
Pipeline Error 1:
out.1.txt
Command output: rsem-extract-reference-transcripts rsem/genome 0 A_thaliana_Jun_2009_genes.gtf None 0 rsem/A_thaliana_Jun_2009.fa "rsem-extract-reference-transcripts rsem/genome 0 A_thaliana_Jun_2009_genes.gtf None 0 rsem/A_thaliana_Jun_2009.fa" failed! Plase check if you provide correct parameters/options for the pipeline!
If the GTF file above can't be used as-is, let's not use it.
Instead, I think we should run pipeline with GTF files from two sources:
- hotpollen/splicing-analysis/ExternalData/Araport11.gtf from https://bitbucket.org/hotpollen/flavonoid-rnaseq (see above, [~aloraine] made it)
- mentioned in previous comment from [~molly] : https://www.arabidopsis.org/download_files/Genes/Araport11_genome_release/Araport11_GTF_genes_transposons.current.gtf.gz
Running pipeline with both, separately, could expose flaws or limitations we would want to know about.
Pipeline Error 2:
out.2.txt
Command error: INFO: Converting SIF file to temporary sandbox... Version Info: ### PLEASE UPGRADE SALMON ### ### A newer version of salmon with important bug fixes and improvements is available. #### ### The newest version, available at https://github.com/COMBINE-lab/salmon/releases contains new features, improvements, and bug fixes; please upgrade at your earliest convenience. ... [2023-04-18 15:58:36.566] [jointLog] [critical] Transcript ATMG01375.1 appeared in the BAM header, but was not in the provided FASTA file [2023-04-18 15:58:36.571] [jointLog] [critical] Please provide a reference FASTA file that includes all targets present in the BAM header If you have access to the genome FASTA and GTF used for alignment consider generating a transcriptome fasta using a command like: gffread -w output.fa -g genome.fa genome.gtf
Compare Araport11 GTF with Tomato GTF to find any formatting issues:
Araport11_GTF_genes_transposons.current.gtf:
Chr1 Araport11 gene 3631 5899 . + . transcript_id "AT1G01010"; gene_id "AT1G01010"; Chr1 Araport11 mRNA 3631 5899 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 CDS 3760 3913 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 CDS 3996 4276 . + 2 transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 CDS 4486 4605 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 CDS 4706 5095 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 CDS 5174 5326 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 CDS 5439 5630 . + 0 transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 exon 3631 3913 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 Araport11 exon 3996 4276 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010";
S_lycopersicum_Jun_2022.gtf:
- Tomato gtf for comparison
- Directory: /nobackup/tomato_genome/scripts/flavonoid-rnaseq/ExternalDataSets/S_lycopersicum_Jun_2022.gtf
0 NA exon 153300 153429 . - . transcript_id "Solyc00T000001.1"; gene_id "Solyc00G000001"; 0 NA exon 159764 159897 . - . transcript_id "Solyc00T000001.1"; gene_id "Solyc00G000001"; 0 NA exon 238088 238260 . + . transcript_id "Solyc00T000002.1"; gene_id "Solyc00G000002"; 0 NA exon 238619 238737 . + . transcript_id "Solyc00T000002.1"; gene_id "Solyc00G000002"; 0 NA exon 240718 242241 . - . transcript_id "Solyc00T000003.1"; gene_id "Solyc00G000003"; 0 NA exon 242297 242691 . - . transcript_id "Solyc00T000004.1"; gene_id "Solyc00G000004"; 0 NA exon 243423 243537 . - . transcript_id "Solyc00T000004.1"; gene_id "Solyc00G000004"; 0 NA exon 242523 242706 . + . transcript_id "Solyc00T000005.1"; gene_id "Solyc00G000005"; 0 NA exon 243393 243590 . + . transcript_id "Solyc00T000005.1"; gene_id "Solyc00G000005"; 0 NA exon 245339 245748 . - . transcript_id "Solyc00T000006.1"; gene_id "Solyc00G000006";
[~aloraine]
TAIR10.gtf (NEW):
- TAIR10 gene models only
- from: https://bitbucket.org/hotpollen/splicing-analysis/src/main/ExternalData/TAIR10.gtf
local aloraine$ grep AT1G01010 TAIR10.gtf Chr1 TAIR10 exon 3631 3913 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 TAIR10 exon 3996 4276 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 TAIR10 exon 4486 4605 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 TAIR10 exon 4706 5095 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 TAIR10 exon 5174 5326 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010"; Chr1 TAIR10 exon 5439 5899 . + . transcript_id "AT1G01010.1"; gene_id "AT1G01010";
Pipeline Successfully ran with the following parameters:
./doIt.sh SRP371294.csv A_thaliana_Jun_2009.fa Araport11.gtf Araport11.bed Arabidopsis.config 1> out.6.txt 2> err.6.txt
Notes: Araport11.gtf worked due to only containing exons and appropriate annotations that were needed. Can only use the command 'wget' on the cluster with open raw data in bitbucket.
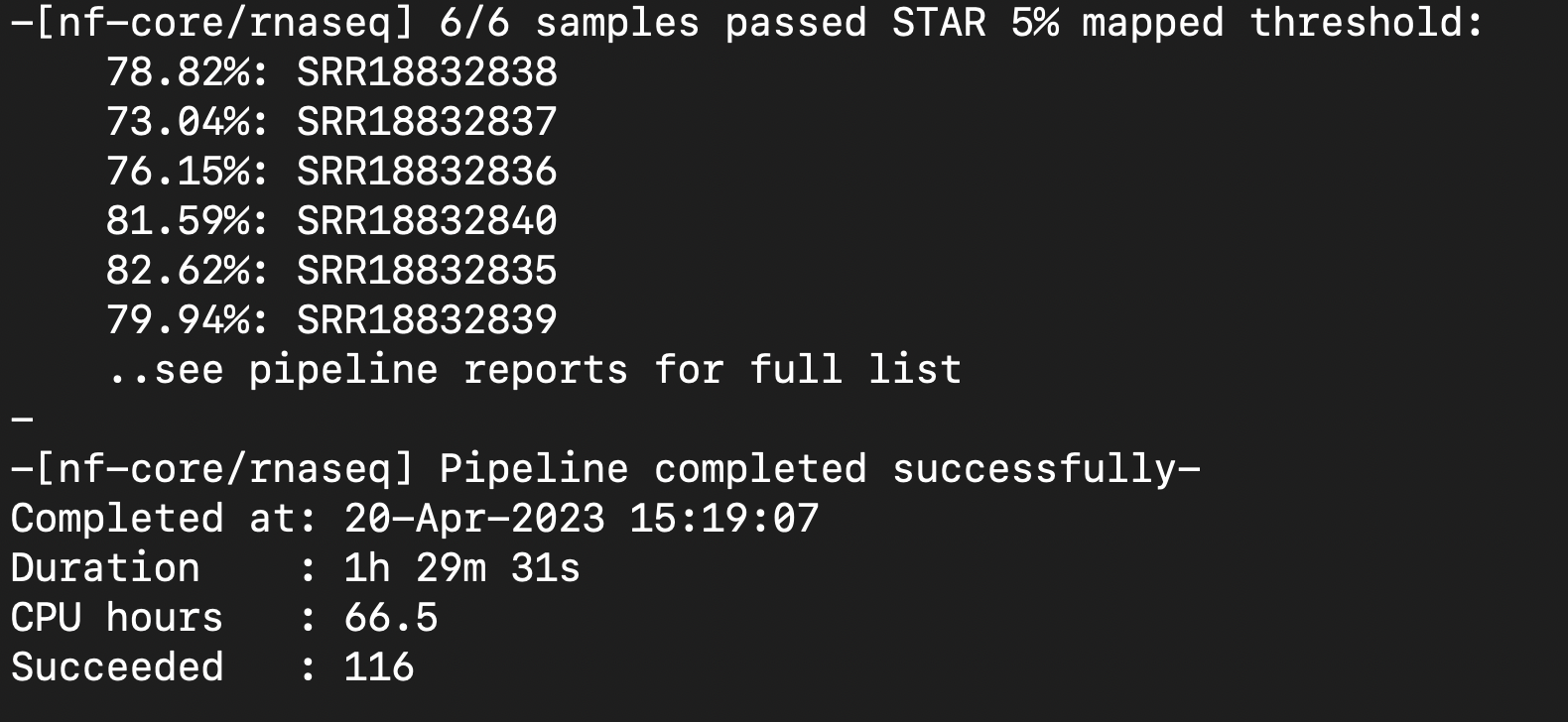
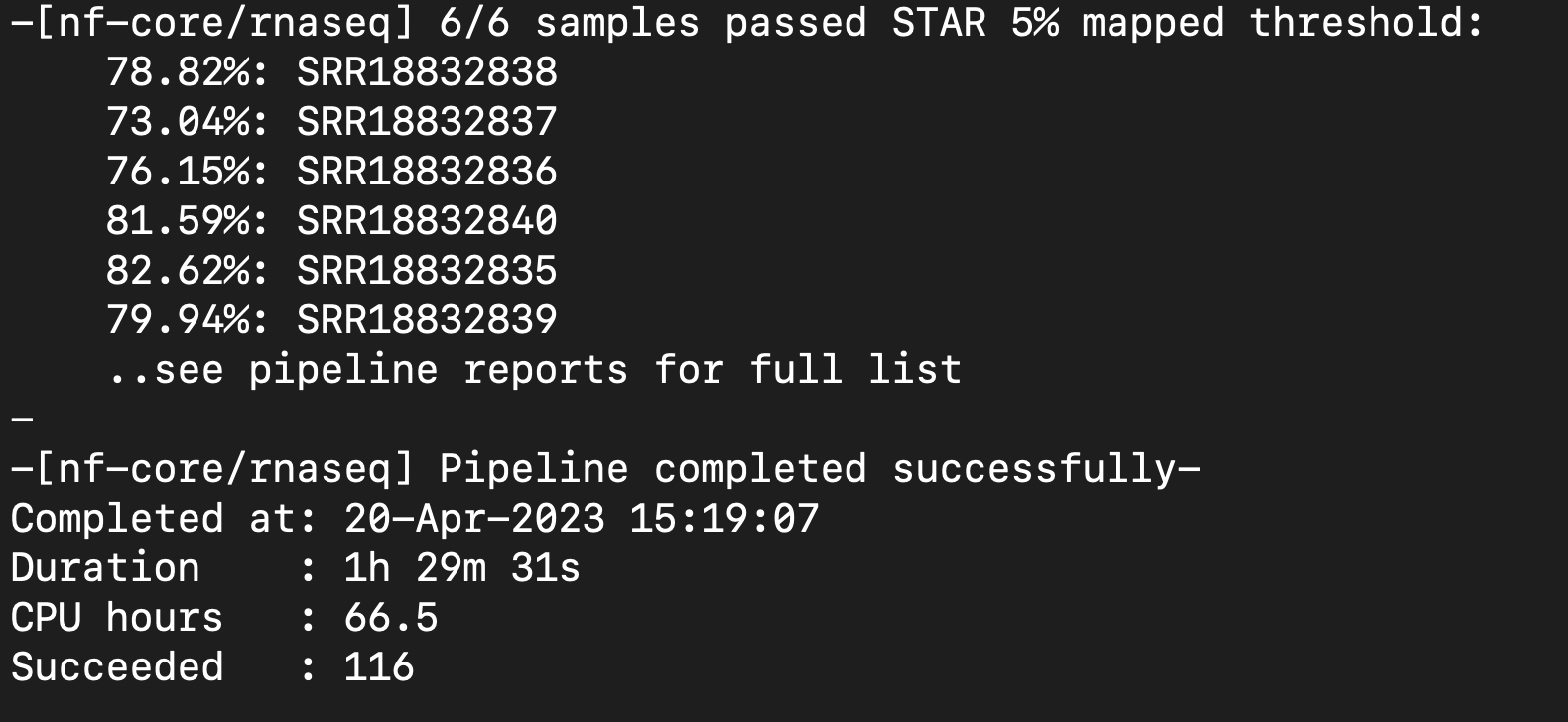
Next Steps: Check multiqc report and determine if strandedness was correct for csv file. Remove the word 'sorted' from bam files. Create coverage graphs and junction files.
scp mdavi258@hpc.uncc.edu:/nobackup/tomato_genome/alt_splicing/SRP371294/results/multiqc/star_salmon/multiqc_report.html ~/Desktop
- Strandedness = unstranded
- Need to fix csv file and rerun pipeline
SRP371294_multiqc_report.html
Fixed Successful Run:
Scaled Coverage Graphs:
./sbatch-doIt.sh .bam bamCoverage.sh >jobs.out 2>jobs.err
- /nobackup/tomato_genome/alt_splicing/SRP371294/results/star_salmon
Junction Files:
- Reference ticket for code
IGBF-3165 - Had to change 2bit file in find_junctions.sh script to 'A_thaliana_Jun_2009.2bit' (see ticket description)
./sbatch-doIt.sh .bam find_junctions.sh >jobs.out 2>jobs.err
- /nobackup/tomato_genome/alt_splicing/SRP371294/results/star_salmon
Review:
- Do I need to add SRP371294.csv, A_thaliana_Jun_2009.fa, Araport11.gtf, Araport11.bed, and Arabidopsis.config to bitbucket? If so, where?
- Do I need to add multiqc report and csv files to bitbucket?
- Do all coverage graphs and junction files work?
- Should I make new directories on cluster for coverage graphs or junction file results?
Request for [~molly]: Please log into the cluster & make sure all the newly made files and directories are group-writable. (So I can make or edit files)
Ok! Let me know if it is working now!
chmod +rwx *
[~aloraine]
Permissions errors:
mkdir: cannot create directory ‘to_transfer’: Permission denied [aloraine@str-i1 SRP371294]$ pwd /nobackup/tomato_genome/alt_splicing/SRP371294
Copied files to /nobackup/tomato_genome/alt_splicing/SRP371294.transfer ( a location where my user has write permission )
Number of files: 36
Number of samples: 6
Size: 15 Gb
Deploying data to:
/projects/igbquickload/lorainelab/www/main/htdocs/rnaseq/A_thaliana_Jun_2009/SRP371294
On RENCI sci-das host.
Note:
This will not be part of the hotpollen Quickload but instead will get put into the "rnaseq" quickload.
Transferring with:
scp -J aloraine@hop.renci.org -r SRP371294.transfer aloraine@lorainelab-quickload.scidas.org:/projects/igbquickload/lorainelab/www/main/htdocs/rnaseq/A_thaliana_Jun_2009/SRP371294/.
Added files to repository:
- SRP371294-multiqc_report.html
- SRP371294-salmon.merged.gene_counts.tsv
- SRP371294.config (copy of /nobackup/tomato_genome/alt_splicing/SRP371294/Arabidopsis.config)
Multiqc file looks fine.
Moving to DONE.
Directory: /nobackup/tomato_genome/alt_splicing/SRP371294
Config parameters from paper that published results(link in description): “–dta –min-intronlen 60 –max-intronlen 6000”
params { modules { 'star_align' { args = '--alignIntronMax 6000 --quantMode TranscriptomeSAM --twopassMode Basic --outSAMtype BAM Unsorted --readF$ } 'hisat2_align' { args = " --max-intronlen 6000 --met-stderr --new-summary --dta" } } }GTF file: https://www.arabidopsis.org/download/index-auto.jsp?dir=%2Fdownload_files%2FGenes%2FAraport11_genome_release
Bed file: Location of bed file?
See: http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/Araport11.bed.gz.
Note that this is a "BED-detail" file and has fields 13 and 14. The nfcore/rnaseq pipeline may not accept this. You may need to modify it to remove fields 13 and 14.
CSV file: Make sure to check mulitqc report to see if strandedness is correct!