Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:3
-
Epic Link:
-
Sprint:Spring 6 2023 Mar 20, Spring 7 2023 Apr 10, Spring 8 2023 Apr 24
Description
This data set from Arabidopsis thaliana contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen.
For this task
- download the data as fastq files from SRA
- align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below)
- for alignment parameters, use original publication (referenced above) and their parameters that they used in their experiment. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species!
- align using same maxIntron parameter reported in the methods section for the paper
- for the above, make a new "config" file
- create coverage graphs
- create junction files
Use this reference genome for alignment:
Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.)
2bitToFa command:
twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa
Attachments
Issue Links
- relates to
-
-
- To-Do
-
Activity
| Field | Original Value | New Value |
|---|---|---|
| Epic Link | IGBF-2993 [ 21429 ] |
| Story Points | 2 | 3 |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen.
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes. For this task, download the data as fastq files and align them against the Arabidopsis TAIR10 genome. Create coverage graphs and junction files, as per usual. Use this reference genome: http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen.
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file and version-control it using name of * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen.
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file and version-control it using name of * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen.
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
| Sprint | Spring 3 2023 Feb 1 [ 163 ] | Spring 4 2023 Feb 13 [ 164 ] |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen.
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
| Rank | Ranked higher |
| Sprint | Spring 4 2023 Feb 21 [ 164 ] | Spring 5 2023 Mar 6 [ 165 ] |
| Sprint | Spring 5 2023 Mar 6 [ 165 ] | Spring 6 2023 Mar 20 [ 166 ] |
| Rank | Ranked higher |
| Assignee | Molly Davis [ molly ] |
| Sprint | Spring 6 2023 Mar 20 [ 166 ] | Spring 6 2023 Mar 20, Spring 7 2023 Apr 10 [ 166, 167 ] |
| Rank | Ranked higher |
| Status | To-Do [ 10305 ] | In Progress [ 3 ] |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} 2bitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check the paper: * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} 2bitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check one of our RNA-Seq data analysis papers that used Arabidopsis TAIR10 genome. Specifically, you need to get the "maximum intron" parameter. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species! * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} 2bitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check one of our RNA-Seq data analysis papers that used Arabidopsis TAIR10 genome. Specifically, you need to get the "maximum intron" parameter. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species! * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} 2bitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check one of our RNA-Seq data analysis papers that used Arabidopsis TAIR10 genome. Specifically, you need to get the "maximum intron" parameter. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species! * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check one of our RNA-Seq data analysis papers that used Arabidopsis TAIR10 genome. Specifically, you need to get the "maximum intron" parameter. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species! * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check one of our RNA-Seq data analysis papers that used Arabidopsis TAIR10 genome. Specifically, you need to get the "maximum intron" parameter. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species! * Use reference genome annotations from the Araport 11 data set * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
| Description |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, check one of our RNA-Seq data analysis papers that used Arabidopsis TAIR10 genome. Specifically, you need to get the "maximum intron" parameter. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species! * Use reference genome annotations from the Araport 11 data set * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
This [data set from Arabidopsis thaliana|https://trace.ncbi.nlm.nih.gov/Traces/?view=study&acc=SRP371294] contains six samples of FACS-sorted sperm and vegetative cells from mature pollen. The publication is here: https://pubmed.ncbi.nlm.nih.gov/36515615/
This would be a useful reference data set for our studies, as the authors reported many differentially and alternatively spliced genes between sperm and vegetative cells harvested from mature Arabidopsis pollen. For this task * download the data as fastq files from SRA * align fastq files using nf-core/rnaseq vs. TAIR10 genome (see link below) * for alignment parameters, use original publication (referenced above) and their parameters that they used in their experiment. Every RNA-Seq to genome alignment tool requires the user to define a maximum intron size parameter. Never use the default! Customize for your species! * align using same maxIntron parameter reported in the methods section for the paper * for the above, make a new "config" file * create coverage graphs * create junction files Use this reference genome for alignment: * http://lorainelab-quickload.scidas.org/quickload/A_thaliana_Jun_2009/A_thaliana_Jun_2009.2bit Create the "fasta" file from the above 2bit file using blat suite tools on cluster. The program you need is 2bitToFa (I think to load it, you have to use "module load blatsuite" or something like that. Use "module avail" to find the correct module name.) 2bitToFa command: {code} twoBitToFa A_thaliana_Jun_2009.2bit A_thaliana_Jun_2009.fa {code} |
| Status | In Progress [ 3 ] | To-Do [ 10305 ] |
| Sprint | Spring 6 2023 Mar 20, Spring 7 2023 Apr 10 [ 166, 167 ] | Spring 6 2023 Mar 20, Spring 7 2023 Apr 10, Spring 8 2023 Apr 24 [ 166, 167, 168 ] |
| Rank | Ranked higher |
| Status | To-Do [ 10305 ] | In Progress [ 3 ] |
| Status | In Progress [ 3 ] | To-Do [ 10305 ] |
| Assignee | Molly Davis [ molly ] | Ann Loraine [ aloraine ] |
| Status | To-Do [ 10305 ] | In Progress [ 3 ] |
| Status | In Progress [ 3 ] | To-Do [ 10305 ] |
| Assignee | Ann Loraine [ aloraine ] | Molly Davis [ molly ] |
| Status | To-Do [ 10305 ] | In Progress [ 3 ] |
| Comment |
[ Pipeline Error 1 solution:
Araport11_GTF_genes_transposons.current.gtf.gz is working with the fasta file. ] |
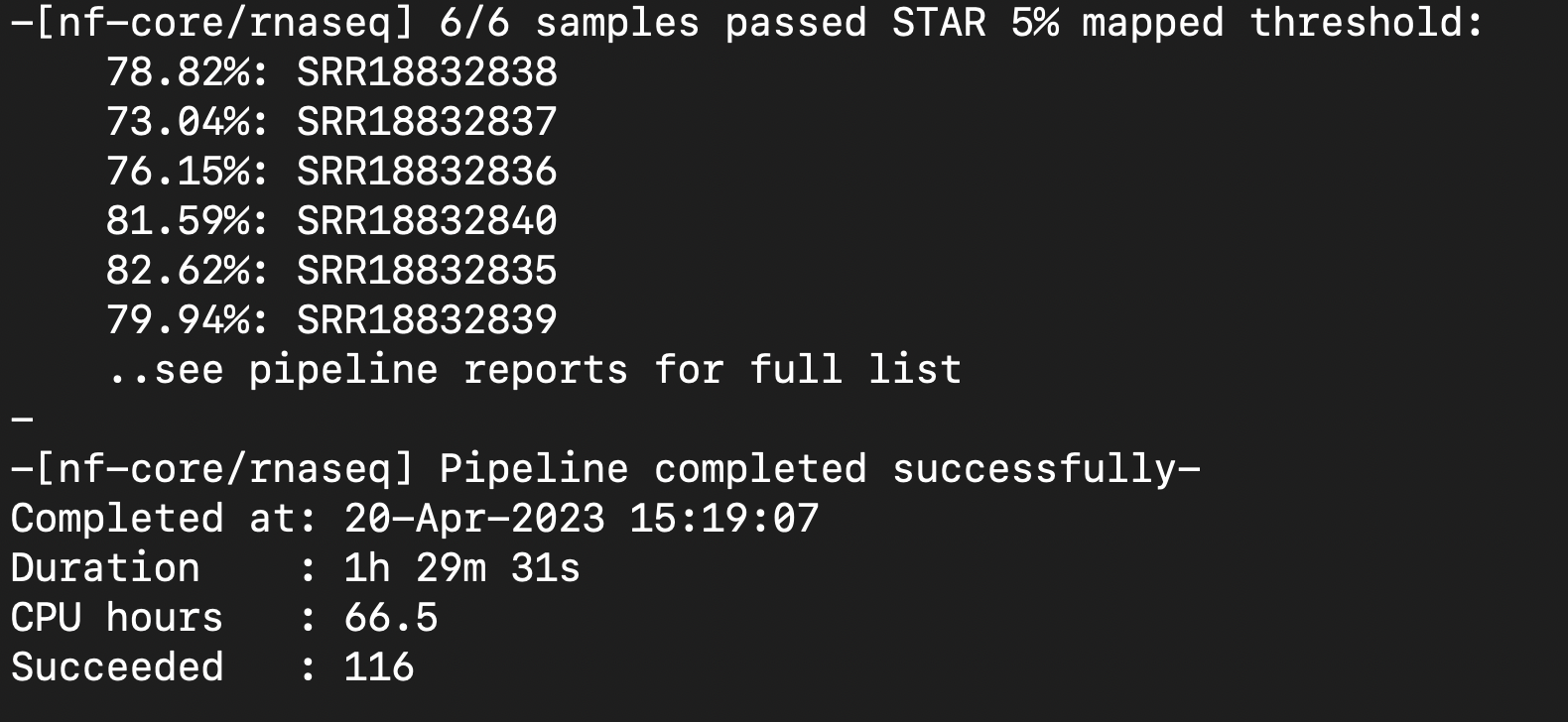
| Attachment | Screenshot 2023-04-20 at 1.32.08 PM.png [ 17864 ] |
| Attachment | Screenshot 2023-04-20 at 1.32.08 PM.png [ 17864 ] |
| Attachment | multiqc_report.html [ 17865 ] |
| Attachment | multiqc_report.html [ 17865 ] |
| Attachment | SRP371294_multiqc_report.html [ 17866 ] |
| Attachment | Successful Pipeline Run 6.png [ 17867 ] |
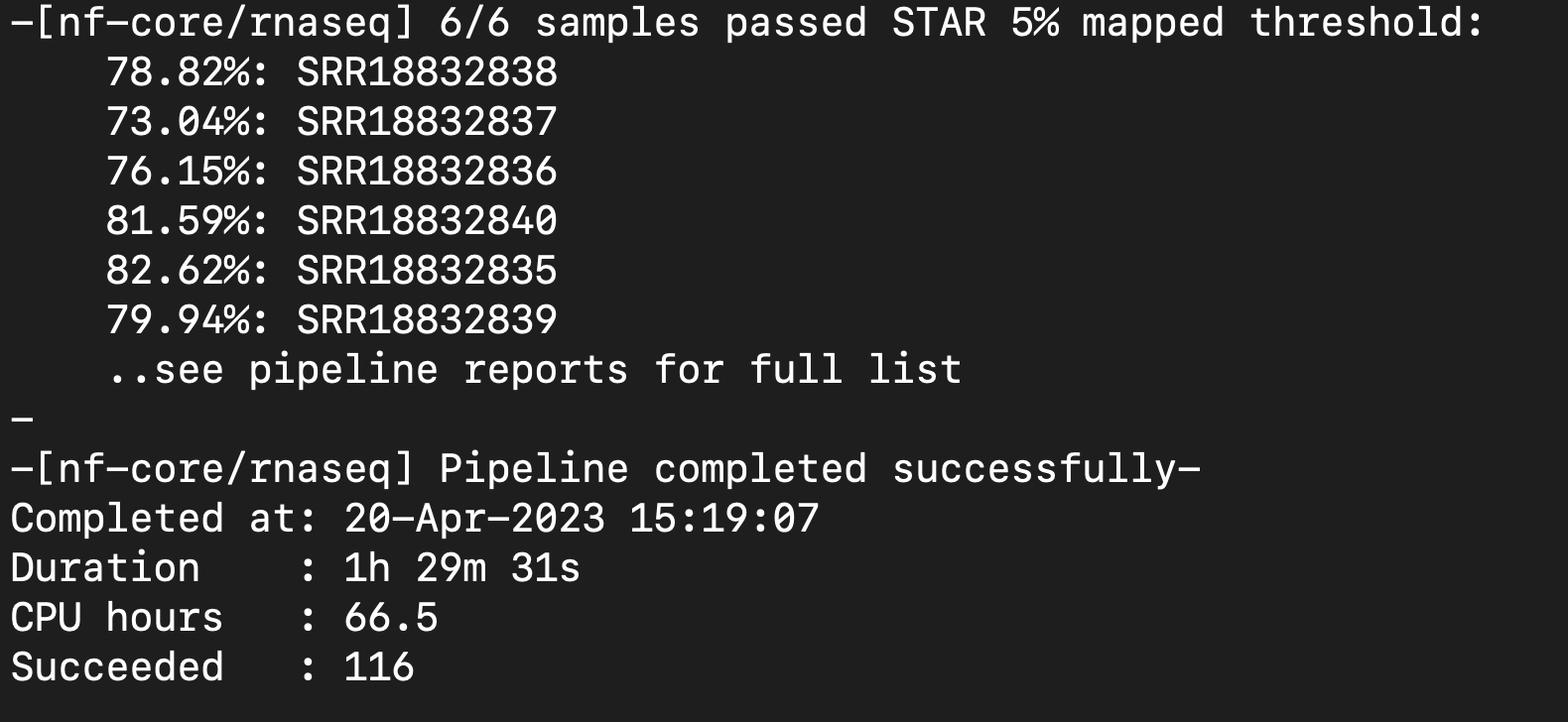{kind=link}
| Status | In Progress [ 3 ] | Needs 1st Level Review [ 10005 ] |
| Assignee | Molly Davis [ molly ] | Ann Loraine [ aloraine ] |
| Status | Needs 1st Level Review [ 10005 ] | First Level Review in Progress [ 10301 ] |
| Status | First Level Review in Progress [ 10301 ] | Ready for Pull Request [ 10304 ] |
| Status | Ready for Pull Request [ 10304 ] | Pull Request Submitted [ 10101 ] |
| Status | Pull Request Submitted [ 10101 ] | Reviewing Pull Request [ 10303 ] |
| Status | Reviewing Pull Request [ 10303 ] | Merged Needs Testing [ 10002 ] |
| Status | Merged Needs Testing [ 10002 ] | Post-merge Testing In Progress [ 10003 ] |
| Resolution | Done [ 10000 ] | |
| Status | Post-merge Testing In Progress [ 10003 ] | Closed [ 6 ] |
| Assignee | Ann Loraine [ aloraine ] | Molly Davis [ molly ] |