Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:3
-
Sprint:Summer 2, Summer 6, Summer 7, Fall 3, Fall 4, Fall 6, Fall 7, Winter 1
Description
Situation: We have identified a publicly available Nebula Genomics CRAM file (link). Nowlan has downloaded the file and created an index, and was able to view the file in IGB. We would like to make this file available to IGB users as an example of a consumer genomics CRAM file.
Task: Upload and deploy the CRAM file (NB72462M.cram), CRAM index, VCF file, and VCF index and make them available through an IGB Quickload. Also deploy the IGBF-3841 via the annots.xml.
Note: Data are from Personal Genome Project
Link to metadata in PGP: https://my.pgp-hms.org/profile/huF7A4DE
Link to PGP search page: https://my.pgp-hms.org/public_genetic_data
Revision:
- Scope increase - include Nebula Genomics and Sequence.com data files for Donor 1
- Scope increase - explore data in region of BRCA1 as preliminary use case, beginning tutorial
Attachments
Issue Links
Activity
Question for Paige Kulzer:
- Did you have to download the entire CRAM file to view it in IGB?
Question for Nowlan Freese:
- Can we make an annots.xml file that points to the remote CRAM file using the third-party provided link and point to the .crai file hosted at another location? I think this might be possible to do with .bai files. I do not know if I am remembering correctly, however. If we can do it with .bai files, it might also be possible with .crai files.
I tried using the link above to open the .cram file in IGB, but that only loaded an empty track. So, yes, the .cram file had to be downloaded so that the .crai file could be created locally before viewing the data in IGB.
Ann Loraine - I'm pretty sure we can specify a separate location for the index file in the annots.xml, but I will double-check and update here. For the third-party provided link, are you referring to the link provided by Nebula genomics? That link is only "live" for a couple of days/weeks before the data are archived and no longer available. Once the data are archived the user has to request the data through their Nebula account. It takes a couple of days before the data are made available again.
reply: No, I am not referring to the Nebula Genomics download link. I meant the link in this ticket to the cram file. If we can refer to it in an annots.xml then we will not have to host it ourselves.
Nowlan - I have the index, I can put it in BioViz Connect and then create an annots.xml and see if I can point it at this cram and the index in BioViz Connect.
Ann Loraine - I was able to create a quickload using the link to the original CRAM file and a link to the CRAM index that I placed in CyVerse (see quickload.zip). Data loaded quickly and I did not see any errors in the log.
From Paige:
I believe you can use samtools to do this! I think this might work: samtools view -Co out.chr1.cram in.cram chr1
To install samtools on a mac, I used Homebrew.
To install Homebrew: /bin/bash -c "$(curl -fsSL https://raw.githubusercontent.com/Homebrew/install/HEAD/install.sh)"
To install samtools: brew install samtools
From Nowlan:
I think the samtools command would be something like:
samtools view -h -C -T GENOME.fa -o NAME_OF_OUTPUT_FILE.cram NAME_OF_CRAM_INPUT_FILE.cram NAME_OF_CHROMOSOME_TO_EXTRACT
samtools view -h -C -T hg38.fa -o nowlan_nebula_chr1.cram nowlan_nebula_full.cram chr1
I'm pretty sure you need the "-T" and the fasta file for the genome associated with the cram file. See the samtools documentation here: https://www.htslib.org/doc/samtools-view.html
Suggestions:
- Use Quickload Builder App to create an Individual Genome Sequencing repository
- It could have two datasets: chromosome 1 from https://e4d05c260dbc25ea5821e03eb009a2b7-26238.collections.ac2it.arvadosapi.com/_/NB72462M.cram
- Chromosome 1 from a more recent Nebula data set
- Document how you did it in a Jira ticket with the intention to transform the ticket comments into one or more publications, e.g., a Bioinformatics Application Note or slidedeck for a talk
- Make one or two postcards with visually appealing images showing off what a personal genome sequence can look like in IGB; hand them out to audience members or others who would be interested in learning about individual genome sequencing
Add the link to the nebula genomics CRAM file to IGB Quickload server (via subversion) in a new folder "Individual genome sequencing".
Can add cram index to repo, add some kind of documentation explaining where the CRAM file came from and additional details. Maybe update readme or add a txt?
- I installed SVN on my new machine, but the subversion repository appears to be down again and is responding with svn: E200029: Couldn't perform atomic initialization
- We had previously discussed some changes to the annots.xml, but I don't think the changes were saved/updated in the ticket (I'm pretty sure the attached zip is the old/original version). I have added an updated version with what I can remember we discussed for review.
- Note that the url listed in the annots.xml will be updated to point at whatever documentation we create about the source of the CRAM file. We will also need to update the location of the index.
I think we should bump this ticket down a couple of sprints. This file will not load in IGB 10.0.1 so we will need to do the IGB 10.1.0 release first. I think it would be confusing for users if a new dataset appeared in the default data for humans but wouldn't load.
Discussed with Dr. Loraine, she suggested adding the changes to annots.xml but having it commented out.
Very sorry for blocking your work. I restarted the svn site and reattached the virtual hard drive storing the data.
Logged in using ec2-user@svn.bioviz.org, then did:
[elastic svn ~]$ sudo su [root@ip-172-31-63-94 ec2-user]# ls /svn [root@ip-172-31-63-94 ec2-user]# mount -t ext4 /dev/xvdf /svn [root@ip-172-31-63-94 ec2-user]# ls /svn genomes lost+found svn-auth [root@ip-172-31-63-94 ec2-user]#
Follow-up regarding suggestion of commentint-out new data sets in Quickload main:
- I am not actually sure IGB can respect comments in an annots.xml file.
Some additional notes:
- Should be able to comment out the new file in the annots.xml (see stackoverflow).
- It looks like we will need to store the index somewhere online that we can provide a full URL to. I have tried multiple versions of the quickload using bam/cram files. The only time the annots.xml index attribute appears to work is if a full URL path is provided for both the name and index attributes. This doesn't line up with our current annots.xml documentation, but does seem to line up with the requirements for the
IGBF-445ticket where the work was done (links to files and their indexes stored online in different locations). Note that the commit for this work was quite extensive, so it is unclear how easy it would be to improve the annots.xml index attribute logic. I also could not find any examples in our current quickloads where we use the index attribute. - See my attached annots.xml for examples of what I tested, the only files that did not throw an error regarding the index attribute were "NB72462M - Personal Genome Project" and "thing4".
Error:
java.lang.IllegalArgumentException: URI is not absolute at java.base/java.net.URL.of(URL.java:862) at java.base/java.net.URI.toURL(URI.java:1172) at org.lorainelab.igb.bam.BAM.getSAMFileReader(BAM.java:106) at org.lorainelab.igb.bam.BAM.init(BAM.java:160) at org.lorainelab.igb.bam.XAM.getChromosomeList(XAM.java:100) at com.affymetrix.genometry.quickload.QuickLoadSymLoader.loadAndAddSymmetries(QuickLoadSymLoader.java:153) at com.affymetrix.genometry.quickload.QuickLoadSymLoader.loadSymmetriesThread(QuickLoadSymLoader.java:139) at com.affymetrix.genometry.quickload.QuickLoadSymLoader.loadFeatures(QuickLoadSymLoader.java:119) at com.affymetrix.igb.view.load.GeneralLoadUtils.loadFeaturesForSym(GeneralLoadUtils.java:768) at com.affymetrix.igb.view.load.GeneralLoadUtils$2.runInBackground(GeneralLoadUtils.java:710) at com.affymetrix.igb.view.load.GeneralLoadUtils$2.runInBackground(GeneralLoadUtils.java:705) at com.affymetrix.genometry.thread.CThreadWorker.doInBackground(CThreadWorker.java:73) at java.desktop/javax.swing.SwingWorker$1.call(SwingWorker.java:305) at java.base/java.util.concurrent.FutureTask.run(FutureTask.java:317) at java.desktop/javax.swing.SwingWorker.run(SwingWorker.java:342) at java.base/java.util.concurrent.ThreadPoolExecutor.runWorker(ThreadPoolExecutor.java:1144) at java.base/java.util.concurrent.ThreadPoolExecutor$Worker.run(ThreadPoolExecutor.java:642) at java.base/java.lang.Thread.run(Thread.java:1583)
Added quickload_v3.zip which includes the IGBF-3841 data.
Update:
- As part of this ticket, I have deployed the full amount of my own data (from Nebula) to this location: https://data.bioviz.org/genomeseq/H_sapiens_Dec_2013/
I started exploring a use case scenario where I visited the gene BRCA1, loaded my data, made a Mismatch pileup graph track, opened IGB Quickload > Genetics and selected Common SNPs (153).
After loading this new Common SNPs track, I zoomed in a bit to get IGB to show the mismatches in the graph and then started looking up my own SNPs.
To do this, I panned from left to right until I found a different-colored vertical stripe in my mismatch pileup graph. Looks like most of the time, the pileup stripe was in the same location as a SNP from the SNP track. I then right-clicked on the SNP and selected option "dbSNP". When I chose that option, my Web browser opened a new page with more information about that SNP. On the SNP page at NCBI, I clicked on a tab labeled "Clinical Significance" to see if that SNP would make me more or less susceptible to breast, ovarian, or other cancers of my reproductive organs.
Use case 2: Looking up a specific SNP
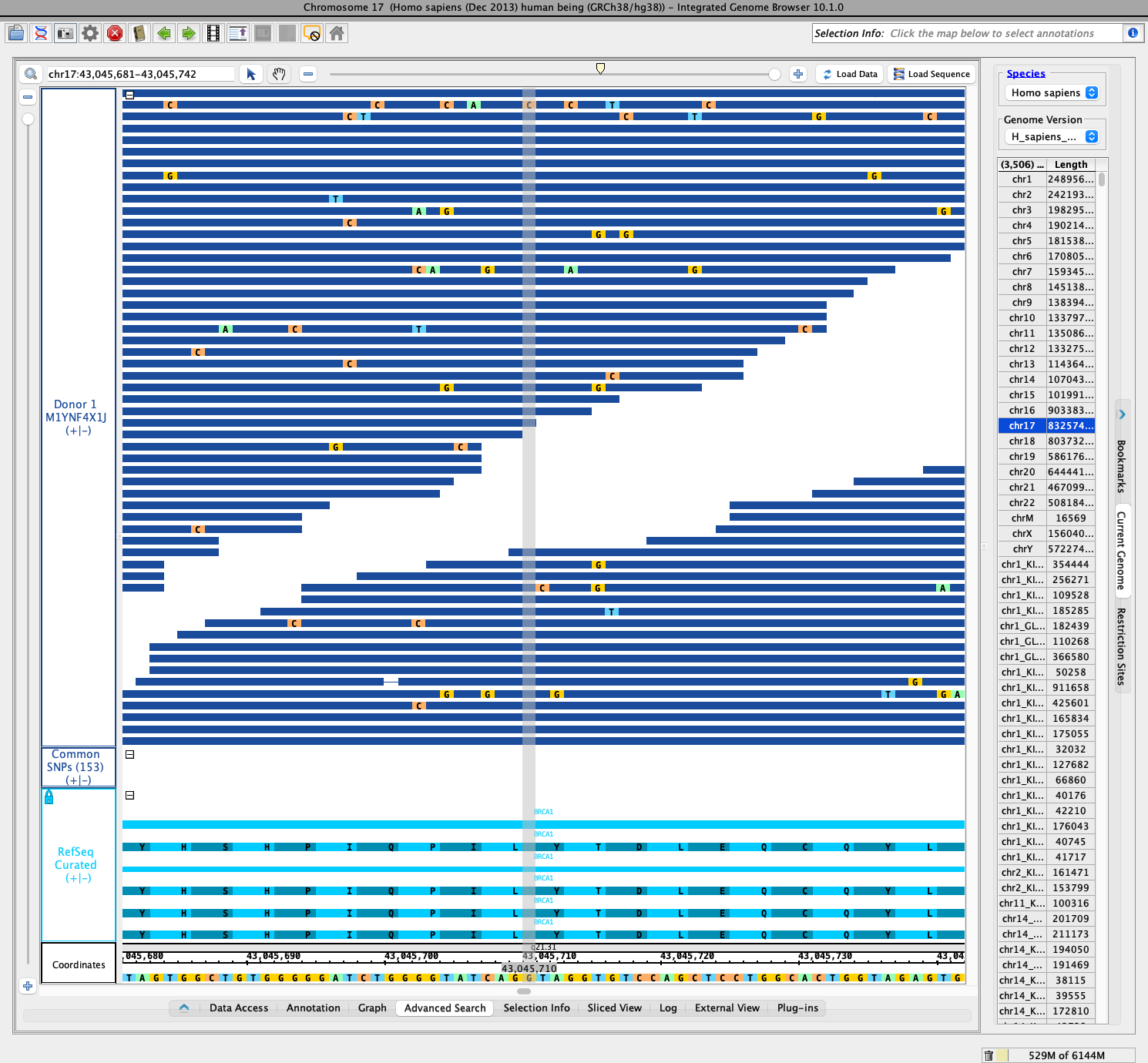
Again, looking at BRCA1, I wanted to focus on SNPs with known cancer risk - e.g., pathogenic SNPs. For this, I tried to use the NCBI Web site to identify a set of regions or variations with known pathogenicity. After some clicking through various pages, I found this page: https://www.ncbi.nlm.nih.gov/snp/rs80357336. This page contains multiple tabs reporting attributes this SNP. The tab named "Clinical Significance" reports that the SNP is "Pathogenic".
In the summary section for the page (above the tabs section), the alleles are reported as "G>A / G>C / G>T" and the position of the SNP in GRCh38.p14 is chr17:43045711. The reference is listed as GRCh38.p14 and, so far as I know, this is the same as H_sapiens_Dec_2013 in IGB.
According to the NCBI page (https://www.ncbi.nlm.nih.gov/snp/rs80357336) the location of the SNP is position 43,045,711. However, the base at this position in the reference, according to IGB, is A, not G. Maybe NCBI is reporting base pair positions using a base numbering system where the first base is numbered 1? In IGB, the first base of a chromosome is numbered 0 - the interbase numbering convention. Base 43,045,710 in IGB is G, which matches the NCBI reference allele, I guess?
I checked the sequences loaded from Donor 1 M1YNF4X1J for IGB base position 43,045,710, and they all match the reference.
I also opened a track for the Common SNPs (153) data set, which comes from dbSNP release 153. I then loaded all the data from this track in the region of BRCA1. However, the SNP of interest - rs80357336 - wasn't among the SNP annotations that got loaded. It is possible this SNP is somewhere else in the genome, but I didn't want to load every annotation from this data because it would likely be too huge, and I know that IGB's search feature will only find items among the previously loaded data. Alternatively, this SNP may not have been present in dbSNP release 153.
In summary, to check a person's genotype for a pathogenic polymorphism, we have to find the pathogenetic polymorphism's location in the reference assembly and then show that location in IGB.
Overall, it seems to be easiest to scan the loaded sequence data looking for differences between the reference and the loaded genome sequence read alignments data and then try to look up whether any differences between the reference and the loaded data correspond to a known pathogenic allele.
I noticed that after loading the Nebula Genomics data for Donor 1 (Nebula sequence), many new sequences appeared in the reference genome assembly "Current Genome" sequence table. After loading Donor 1's data, there were 3,506 sequences listed. However, when I first opened the hg38 genome, there were only 595 sequences listed!
A lot of these had names including strings "decoy" and "HLA".
After searching the web using text "what are decoy sequences in human genome alignment", I found this page at BioStars: https://www.biostars.org/p/73100/#73167.
According to one person's answer (39 upvotes):
The reference genome is incomplete, particularly around the centromeres, so often reads which truly belong elsewhere are wrongly mapped to a particular place in the genome because the true match is missing from the reference. These cause false positive calls, which were bothering us in the 1000 Genomes Project. The decoy is a pragmatic solution to this - it contains known true human genome sequence that is not in the reference genome, and will "suck up" reads that would otherwise map with low quality in the reference. The decoy was built by Heng Li, at the Broad, working with Richard Durbin (Sanger) and Deanna Church of the Genome Reference Consortium (who maintain the reference genome).
Donor 1 BAM file from Sequencing.com also contains these decoy sequences.
Since these "decoy" sequences get included in the alignment process to avoid false positive alignments, probably these should not get shown to users in IGB.
Also, I noticed that they do not show up when I opened and loaded data from Donor NB72462M. However, when I loaded data from this person, a new sequence appeared in the list named "chrEBV". Probably this is Epstein Barr viral genome? This sequence is not present in our 2bit file for hg38, however. When I tried to load the data for this sequence from the NB72462M CRAM-format data file I observed an error in the IGB log. Because IGB can only show alignments from CRAM files when it has access to the reference genome sequence for the alignments, it is not suprising that IGB reports an error and no data can be loaded. For reference, I am including a report from the log - see attached.
However, even after this error, I was able to navigate to other locations in the genome and load reads. So, the error is not a show-stopper!
Update:
Copied data files SQ867JX4-30x-WGS-Sequencing_com-12-25-24.1.fq.gz and SQ867JX4-30x-WGS-Sequencing_com-12-25-24.2.fq.gz to /projects/tomato_genome/fnb/dataprocessing/human_genome in preparation for possible re-alignment of the data.
I think this might be a good idea because the reference genome sequence in the .bam alignment files is unknown. The chromosome names do not match hg38 in that chromosome numbers are used without the chr prefix. It's possible this won't matter and that I'll be able to open and visualize BAM data in IGB thanks to IGB's built-in synonyms file - see: core/synonym-lookup/src/main/resources/chromosomes.txt.
However, I see that we do not have chromosome synonyms for every numbered chromosomal sequence, e.g., chr21_GL383581v2_alt.
For Donor 1's Nebula data, I can "clean up" the alignments files by running samtools view with a reference genome option, using the hg38 reference genome only. If this option works the way I expect, this will allow me to create a new alignment file that eliminates alignments to sequences that are not in the reference.
Also, I noticed that the header information for Donor 1's Nebula data is very verbose. The "CL" annotation is very long, and all that information appears to be getting loaded into IGB for every single alignment shown in the track.
Similarly, I would like to "clean up" the headers. The samtools view command I have in mind for getting rid of the decoy alignments might help if the CL tag gets replaced with a new on. Not sure about that.
Update:
- I loaded both Donor 1 files - the one from Nebula and the one from Sequencing.com
- After loading them, the Current Genome table listed 6,062 sequences!
- I was able to look at data from both files for the canonical reference sequences, however, thanks to IGB's on-board chromosomes.txt files
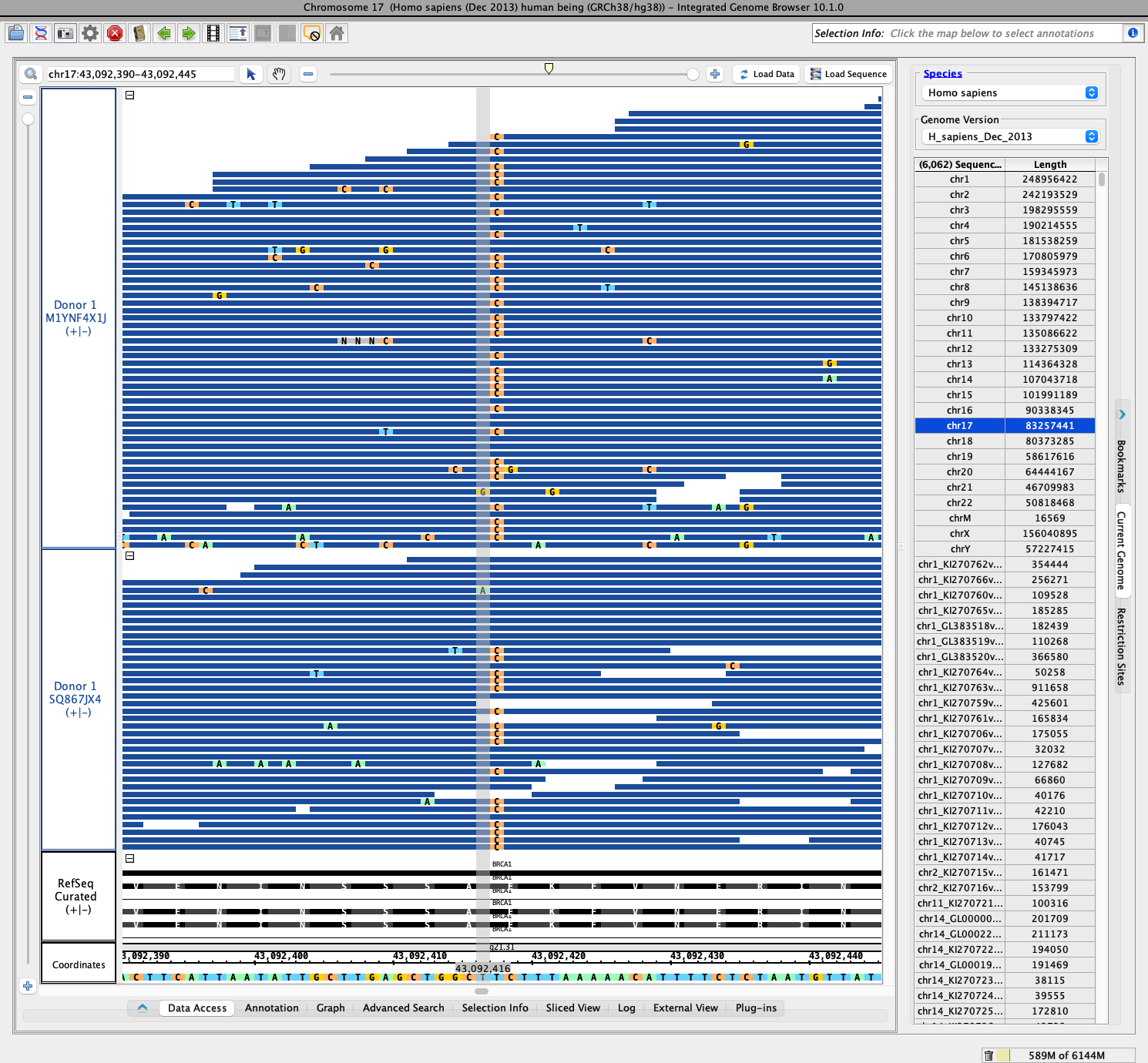
- I checked a location in BRCA1 and noticed that both files contained the same polymorphism and that about half of the reads contained the non-reference allele, indicating Donor 1 is heterozygous in this position
- I made a PNG for the location for future reference and attached it to this ticket
Summary:
Added the following text to H_sapiens_Dec_2013/annots.xml:
<file name="https://data.bioviz.org/genomeseq/H_sapiens_Dec_2013/M1YNF4X1J.cram"
index="https://data.bioviz.org/genomeseq/H_sapiens_Dec_2013/M1YNF4X1J.cram.crai"
title="Genetics/Individual/Donor 1 M1YNF4X1J"
description="Donor 1 whole genome sequencing read alignment results from Nebula Genomics"
direction_type="none"
name_size="14"
max_depth="10"
background="FFFFFF"
foreground="1B4C9B"
show2tracks="false"
/>
<file name="https://data.bioviz.org/genomeseq/unknown_reference/SQ867JX4-30x-WGS-Sequencing_com-12-25-24.bam"
index="https://data.bioviz.org/genomeseq/unknown_reference/SQ867JX4-30x-WGS-Sequencing_com-12-25-24.bam.bai"
title="Genetics/Individual/Donor 1 SQ867JX4"
description="Donor 1 whole genome sequencing read alignment results from Sequencing.com"
direction_type="none"
name_size="14"
max_depth="10"
background="FFFFFF"
foreground="1B4C9B"
show2tracks="false"
/>
<file name="https://e4d05c260dbc25ea5821e03eb009a2b7-26238.collections.ac2it.arvadosapi.com/_/NB72462M.cram"
index="https://data.cyverse.org/dav-anon/iplant/home/nowlanf/cram/index/NB72462M.cram.crai"
title="Genetics/Individual/NB72462M - Personal Genome Project"
description="Whole genome sequencing read alignments from participant huF7A4DE file NB72462M.cram"
direction_type="none"
name_size="14"
max_depth="10"
background="FFFFFF"
foreground="1B4C9B"
show2tracks="false"
url="https://my.pgp-hms.org/profile/huF7A4DE"
/>
Committed change to annots.xml file, incrementing subversion repository to revision 207.
Deployed changes to:
- Quickload hosted at RENCI - Web address http://igbquickload-main.bioviz.org/quickload, file system location /projects/igbquickload/lorainelab/www/main/htdocs/quickload
- Quickload hosted at UNC Charlotte - Web address http://igbquickload.org/quickload, file system location /mnt/igbdata/quickload
To update, I logged into the respective hosts, navigated to the location of the checked-out repository, and executed command:
svn up
For developing the site, I modified a local copy of the Quickload repository, with all the default (factory setting) Quickloads de-activated.
Then, I checked in the changes and deployed the new content to the hosts, as noted in the preceding comment.
To check that the changes are properly deployed, I did the following:
- Launched IGB version 10.1.0, downloaded from the BioViz.org Web site (this is the "released to the public" version of the software)
- Reset IGB Preferences to defaults
- Re-launched IGB version 10.1.0
- Clicked the Mona Lisa image to load genome version H_sapiens_Dec_2013, also known as GRCh38 and hg38.
- Opened Available Data folder named IGB Quickload
- Observed that the Genetics folder now contains sub-folder "Indvidual" containing three items
- Confirmed there are now two checkboxes for Donor 1, neither of which has a link-out icon, as intended
- Also confirmed there is a new checkbox for NB72462, with a linkout icon. Confirmed that when I click the linkout out icon, a page hosted at the Personal Genome Project Web site opens, describing the donor of these data
- Selected each checkbox to open these datasets.
- Noted that all three are using a darkish-blue foreground color, as intended
- Navigated to BRCA1 gene, by entering the gene name in the Advanced Search tab (Properties Search, whole genome) - BRCA1 (note that the Quick Search will take you to a different gene). After running the search, double-click one of the rows with title field listed as the gene name - BRCA1
- Zoomed into the middle long exon by double-clicking it in the display (for your reference, the location is chr17:43,091,263-43,095,031)
- Clicked button "Load Data"
- Observed that all three tracks now have data in them. Also, observed that IGB automatically loaded sequence data for the region, but not for the entire gene - this is expected.
- Observed that the number of sequences listed in the Current Genome tab increased to 6,062, as expected, thanks to the "decoy" sequences and lack of chromosome synonyms problems described in the previous comments. This is expected, but we will probably try to fix this later in future issues. (For example, we could deploy a new chromosomes.txt file via IGB Quickload to fix extra chromosomes getting added to the display from the Donor 1 SQ867JX4 dataset and we could add the Epstein Barr virus sequence to our 2bit file. Finally, we could modify the Donor 1 files to remove the "decoy" alignments.
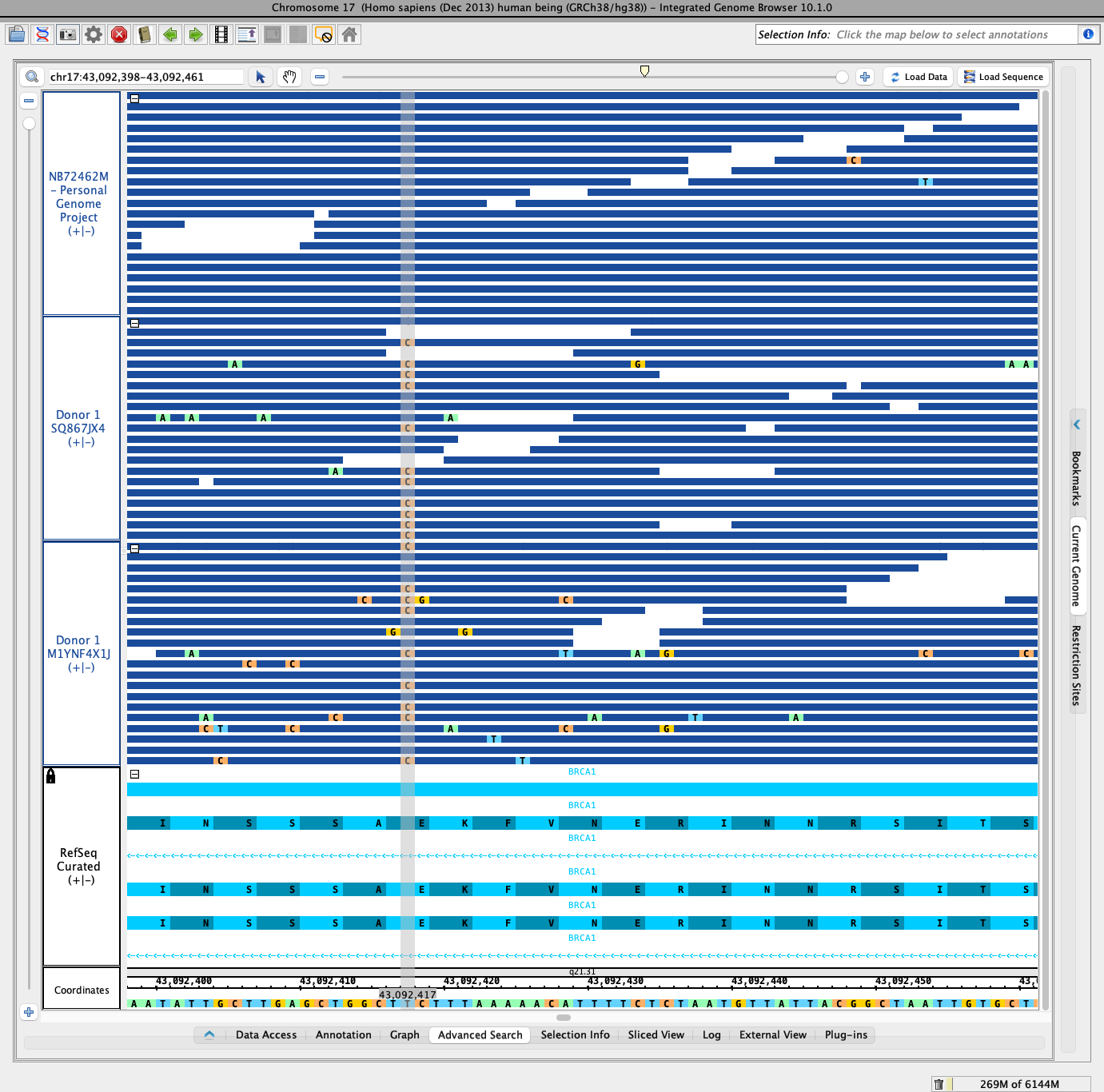
- Observed there is a polymorphism at position chr17:43,092,417. The polymorphism appears in about half of the aligned reads in the two Donor 1 tracks and in none of the aligned reads in the NB72462M track.
To make the image more interesting and appealing, and useful for training, I also did the following:
- Combined RefSeq track's plus and minus strands into a single track. Note that the gene is on the minus strand, so transcription proceeds from right to left in the IGB display.
- Locked the RefSeq track height to 250 pixels to ensure there is enough vertical space to show the amino acid translations for individual codons.
- Expanded the RefSeq track so that the annotations take up all available vertical space. For this, I set the track height to 5, so that the fifth item in the stack occupies the "slop row" at the top of the track.
- Changed the foregroud color of the RefSeq track to aqua so that it will be easier to distinguish adjacent codons and their amino acid translations.
- Changed the track label foreground of the RefSeq track to black to improve readability.
- Loaded genomic sequence for the entire gene to ensure that IGB shows the amino acid residues on top of the gene models. For this, I double-clicked on a gene model to zoom out to the entire gene region, and then clicked "Load Sequence."
- Zoomed back in to the polymorphism in that middle exon.
- Selected all three genome sequence tracks and changed their stack heights so that all read alignments are shown in their own sub-row. Observed that this caused the reads to be crushed too much, making it impossible to see the individual bases of the reads. So, then changed the stack height to 20 for each track.
- Retracted the side and bottom tabbed panel trays to make more room.
- Deselected the genome sequence tracks.
- Made a photo of the scene - see attached file named BRCA1-chr17-43092417.png
Suggestions for testing usability, usefulness and functionality:
- Use NCBI Web site (or other sources) to identify a pathogenic cancer-causing polymorphism in BRCA1
- Check the genotype of each sample. Do Donor 1 and/or Personal Genome Project participant huF7A4DE have the pathogenic allele of this polymorophism?
By trying to do this task, you will gain insight into what individuals will likely want to do when exploring their own data.
To test this ticket, I decided to use ClinVar to identify a pathogenic cancer-causing single nucleotide variant in BRCA1.
I first added ClinVar as a custom web link in IGB by clicking Tools > Configure Web Links and then clicking the "Create New" button and entering the following URL Pattern: https://www.ncbi.nlm.nih.gov/clinvar/?term=$$
Unfortunately, when I right-clicked on an SNV at chr17:43,093,219 and chose to search ClinVar with the new web link I made, this returned no results because the ID being searched (FP270005330L1C046R03305352005) is not recognized by ClinVar. And, there was no obvious way to query ClinVar by a position in the chromosome using bp coordinates, so I couldn't look up the SNV that way, either.
Back in IGB, I then right-clicked on the gene model at that location and used the new ClinVar web link to search "BRCA1" which returned lots of results, though they were non-specific to the point in the genome I clicked.
I looked through the search results and found a well-reviewed pathogenic SNV on the BRCA1 gene at the following location: chr17:43,045,711. There were no polymorphisms present in either of the three donor tracks. I repeated this process several times with various SNV's as well as pathogenic insertions and deletions but found none present in the donor tracks.
Overall, I think there will be a bit of friction for new users that want to use external databases to cross-reference their polymorphisms directly from IGB, but there is a lot of potential now to update our training in a way that gets people started using IGB to view their personal genomics data by using this Quickload.
Re today's quick discussion during the scrum:
Thank you Paige Kulzer for the review and comments!
If you notice anything about how the data are deployed on the web site that is hosting the data, maybe make a new ticket for that and put it into the next sprint. Then, you could close this ticket if you see fit!
Now that I've taken a closer look at how the data are deployed on the web site that is hosting the data, I've discovered that Donor 1 SQ867JX4 is being loaded on the wrong genome. The data for this donor is being stored in a folder called unknown_reference, but, per the text file in that folder (ULTIMATE-COMPATIBILITY-SQ867JX4-30x-WGS-Sequencing_com-12-25-24.txt), this donor's raw genotype data was aligned to GRCh37, not GRCh38:
[...] We are using reference human assembly build 37 (also known as Annotation Release 104). [...] More information on reference human assembly builds: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/
I'm going to make a new ticket to address this issue and will now close this one!
Wow - thanks Paige Kulzer for catching this!!!!
I've confirmed that I can visualize this CRAM data in IGB 10.1.0 (although the VCF file doesn't seem to be loading). Although it's unclear if Nebula Genomics was the genome re-sequencing company that produced this data, I think we should move forward with deploying it and making it available as an IGB Quickload ahead of the ASHG conference this Fall.
Passing this ticket to Nowlan Freese for next steps.