Details
-
Type:
Bug
-
Status: Closed (View Workflow)
-
Priority:
 Blocker
Blocker
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: 9.1.0 Major Release
-
Labels:
-
Story Points:2
-
Epic Link:
-
Sprint:Winter 2018 Sprint 3, Fall 2019 Sprint 1, Fall 2019 Sprint 2, Fall 4 : 30 Sep to 11 Oct, Fall 5 : 14 Oct to 25 Oct, Fall 6 : 28 Oct to 8 Nov
Description
Working with PacBio long reads from a URL, hit a SocketTimeoutException. The reads do not display correctly - appearing as unfilled squares. The read scores also seem to be incorrect, though that may be a separate issue.
Example bam can be found here
Attachments
Activity
This may be related: https://jira.transvar.org/browse/IGBF-1173
Task:
- Find out what is causing the issue
- Propose a fix
Note: The files may have a problem. To check them, you can use samtools command line tool.
Example BAM files:
View BAM file header:
samtools view -H http://igbquickload.org/rnaseq/H_sapiens_Dec_2013/SRP056969/SRR1957124_Adrenal_gland.bam
Get data and header in binary format for a region and redirect to a local file:
samtools view -hb http://igbquickload.org/rnaseq/H_sapiens_Dec_2013/SRP056969/SRR1957124_Adrenal_gland.bam chr11:10,304,780-10,307,600 > ADM.bam
Make index for above file:
samtools index ADM.bam
View data in above file as plain text not binary:
samtools view ADM.bam
Pulled down a small region from the Genome in a Bottle pacBio data (attached as pacBio.bam). Data are from chr1:9,747-20,516.
Note: the pacBio.bam header specifies the first chromosome as "1" instead of "chr1".
Data load in IGB 9.1 (as of Oct. 16, 2019) and 9.0.2 release, but the base sequence does not show correctly. However, no errors are shown in the console. Additionally, the soft-clipping behavior is a little odd - soft-clips show correctly (including the bases), but when hide soft-clipping is selected the soft-clipping sections of the reads are not hidden correctly.
I have attached the reads from SAMTools. I tried to view the pacbio file using Samtools, the data looks fine but the parsing is not happening properly.
Need to investigate more on that
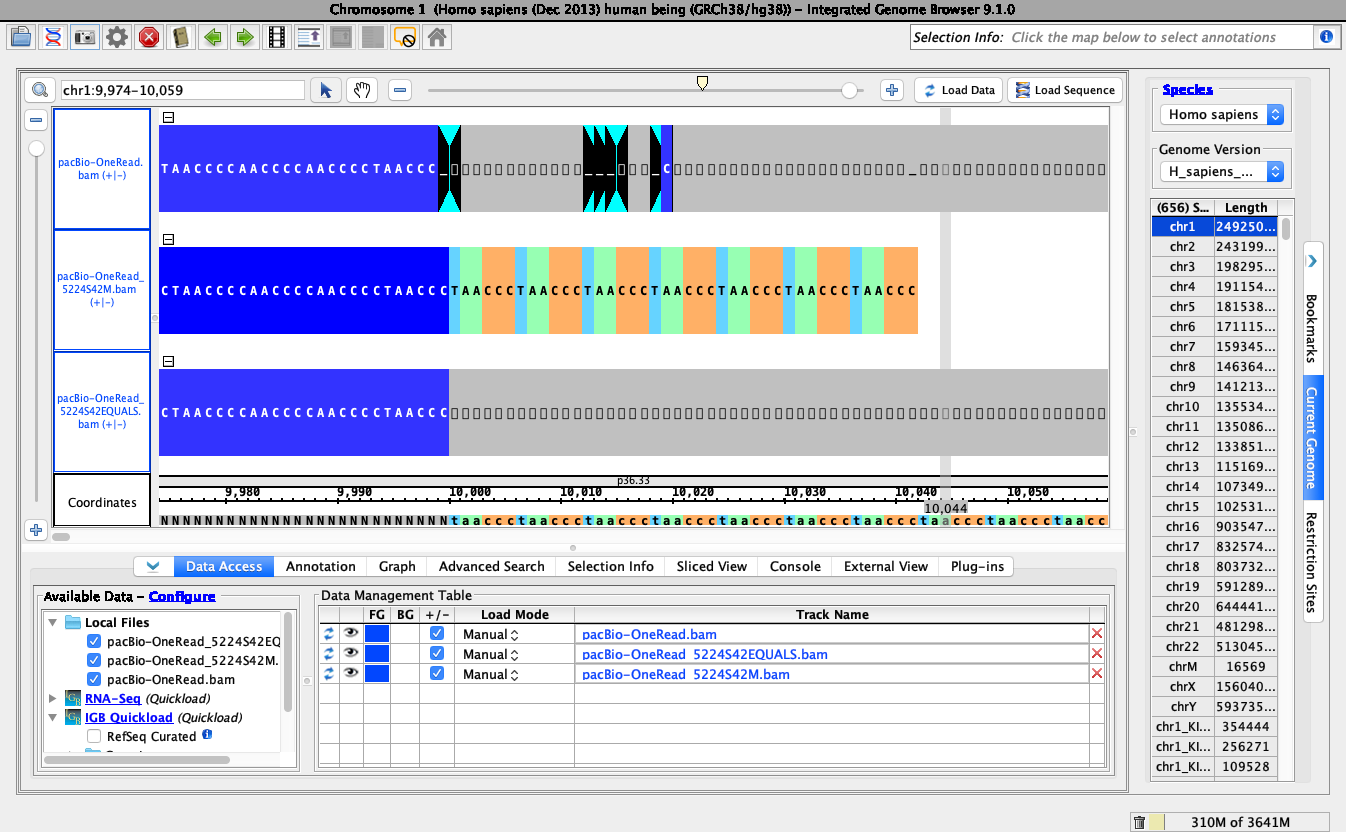
Dr. Loraine's comment about the CIGAR string not being parsed looks to be exactly correct. I've attached several bam files where each file has only a single read. In two of the bam files I've removed all of the read bases except for the beginning soft-clipped part and the first matching bases (5,224 soft clipped followed by 42 matching bases). The difference between the two files is the use of the CIGAR M (alignment match) versus CIGAR = (sequence match). In the image (attached) comparing the three files, when the CIGAR = is used, IGB does not show the read correctly, but by changing the CIGAR to M, IGB shows the read correctly.
I'm not exactly sure the difference between CIGAR M and CIGAR =. Either way, it appears that IGB is having difficulty parsing this particular CIGAR (may also be having difficulty with X (sequence mismatch). I think the next step in this ticket should be to investigate CIGAR parsing in IGB.
Edit:
CIGAR M indicates either a match or mismatch (does not differentiate) at the basepair location. The CIGAR = specifies a match and X specifies mismatch. Whether M or =/X is used most likely depends on the aligner.
The issue is in the BAMSym.java file in the interpretCigar method.
We are not handling the = or X scenarios. I think we can just add in both of them and use the same logic we have for the CIGAR M. The logic for showing/hiding mismatches is handled elsewhere in IGB, so we should be able to treat =/X exactly as M.
I've added some additional files for testing that contain just = or X reads.
Noor and I tested the changes to BAMSym.java, but there were still some issues.
There are two other files that make calls to CigarOperator: XAM.java and VCF.java
It looks like XAM.java is also only handling the M cigar, so probably need to apply the same logic to X/=.
I don't think we need to make changes to the VCF.java. The logic is different and would not apply to the issue we are seeing in bam files.
Reviewed on Noor's branch. Tested using pacBio.bam and pacBio-oneRead.bam which contain CIGAR operators with X/= and softclipping.
Reads are showing correctly. Selection info appears correct. View Read Sequence appears correct for read and softclip. Depth Graph All, Depth Graph Start, and Mismatch Graph appear correctly.
Softclips appear correctly, changing color or hiding works correctly.
Ready for pull request.
Tested using pacBio.bam file.
Data appear correctly in IGB, softclipping appears correctly, and track operations appear to be working correctly.
Closing issue.
This issue may not have anything to do with the reads themselves, and may be an issue with the data being pulled from an ftp site. However, when I pull Illumina short reads from the same site, they load in IGB normally. We haven't tested long reads very much in IGB (PacBio are around 10,000 bp long, versus Illumina 150 bp long), and I'm wondering if the long reads are behaving correctly.