Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:1
-
Epic Link:
-
Sprint:Fall 9 : 9 Dec to 20 Dec, Spring 1 : 25 Dec to 17 Jan
Description
The bedgraph is from estimate coverage. Compare it to the bai file loaded in IGB. (Human 2009 genome).
Attachments

Issue Links
Activity
I agree with this proposed solution.
As a sanity check, I tested an unsorted and scrambled sam file to see how samtools sort would behave.
The starting sam file (unsorted.sam) is:
@SQ SN:chr3 LN:198022430
@SQ SN:chr15 LN:102531392
@SQ SN:chr1 LN:249250621
SOLEXA-1GA-2_2_FC20EMB:5:19:384:574 0 chr1 540627 25 36M * 0 0 AAGGGGCGGCCTGTGGCGTTTTCCTGTAAAGTTGGG hhThhhVa\XOh_JVFDLLQZGCEDFE?B?DGAA>C NM:i:0 X0:i:1 MD:Z:36
SOLEXA-1GA-2_2_FC20EMB:5:3:835:634 0 chr1 159002 25 36M * 0 0 ACACAAACACACAAACACACACACACACAACCCCAA ^ThgYWLRQNNWNHLQGMDIHIFLFKGI?DHAEDA@ NM:i:1 X1:i:1 MD:Z:13A15A6
SOLEXA-1GA-2_2_FC20EMB:5:123:425:446 0 chr15 22809005 25 36M * 0 0 GTGCAGTCCACCAGCAAGGGAGCTGTGACGAGAGTA ]K\HLJGGFLGEIEB?FIBFEIDCC@>EA>@??>=? NM:i:1 X1:i:1 MD:Z:28C3A2A0
SOLEXA-1GA-2_2_FC20EMB:5:241:482:157 0 chr15 22691475 25 36M * 0 0 TCAGTGATAGAGCAAGAAAACAAAATGGGTTTCCTG hhhhhhhhhhbhafWeXW]TOLTHUTVTWHWUK@MC NM:i:0 X0:i:1 MD:Z:36
SOLEXA-1GA-2_2_FC20EMB:5:118:97:121 0 chr3 870368 25 36M * 0 0 TAGCATAGAGTGGTATAACACATTTAAGTATGAAAG hhhhhhhhhhhhhhhhchhhhOhhhYZhcc[TJWCS NM:i:1 X1:i:1 MD:Z:0T35
SOLEXA-1GA-2_2_FC20EMB:5:44:973:514 0 chr3 682558 25 36M * 0 0 CACACACATACACATACATACACACCAACCAACACA hhghhhhhhh]h`ehh\YdXd`haVQKPKJHMEGFF NM:i:1 X1:i:1 MD:Z:20C4C3C6
Note that the header is in the order chr3, chr15, chr1, the reads are in the order chr1, chr15, chr3, and the reads are not positionally sorted.
I ran the following commands (samtools version 1.9):
samtools view -bh unsorted.sam > unsorted.bam
samtools sort
samtools sort -o sorted.bam unsorted.bam
samtools index sorted.bam
samtools view -h sorted.bam > sorted.sam
The output sam file (sorted.sam) is:
@HD VN:1.6 SO:coordinate
@SQ SN:chr3 LN:198022430
@SQ SN:chr15 LN:102531392
@SQ SN:chr1 LN:249250621
SOLEXA-1GA-2_2_FC20EMB:5:44:973:514 0 chr3 682558 25 36M * 0 0 CACACACATACACATACATACACACCAACCAACACA hhghhhhhhh]h`ehh\YdXd`haVQKPKJHMEGFF NM:i:1 X1:i:1 MD:Z:20C4C3C6
SOLEXA-1GA-2_2_FC20EMB:5:118:97:121 0 chr3 870368 25 36M * 0 0 TAGCATAGAGTGGTATAACACATTTAAGTATGAAAG hhhhhhhhhhhhhhhhchhhhOhhhYZhcc[TJWCS NM:i:1 X1:i:1 MD:Z:0T35
SOLEXA-1GA-2_2_FC20EMB:5:241:482:157 0 chr15 22691475 25 36M * 0 0 TCAGTGATAGAGCAAGAAAACAAAATGGGTTTCCTG hhhhhhhhhhbhafWeXW]TOLTHUTVTWHWUK@MC NM:i:0 X0:i:1 MD:Z:36
SOLEXA-1GA-2_2_FC20EMB:5:123:425:446 0 chr15 22809005 25 36M * 0 0 GTGCAGTCCACCAGCAAGGGAGCTGTGACGAGAGTA ]K\HLJGGFLGEIEB?FIBFEIDCC@>EA>@??>=? NM:i:1 X1:i:1 MD:Z:28C3A2A0
SOLEXA-1GA-2_2_FC20EMB:5:3:835:634 0 chr1 159002 25 36M * 0 0 ACACAAACACACAAACACACACACACACAACCCCAA ^ThgYWLRQNNWNHLQGMDIHIFLFKGI?DHAEDA@ NM:i:1 X1:i:1 MD:Z:13A15A6
SOLEXA-1GA-2_2_FC20EMB:5:19:384:574 0 chr1 540627 25 36M * 0 0 AAGGGGCGGCCTGTGGCGTTTTCCTGTAAAGTTGGG hhThhhVa\XOh_JVFDLLQZGCEDFE?B?DGAA>C NM:i:0 X0:i:1 MD:Z:36
Note that the header remains in the order chr3, chr15, chr1, after sorting the reads appear in the order chr3, chr15, chr1 and are positionally sorted.
This provides further evidence that there is no default sort order for the chromosomes when using samtools. The header determines the chromosome order.
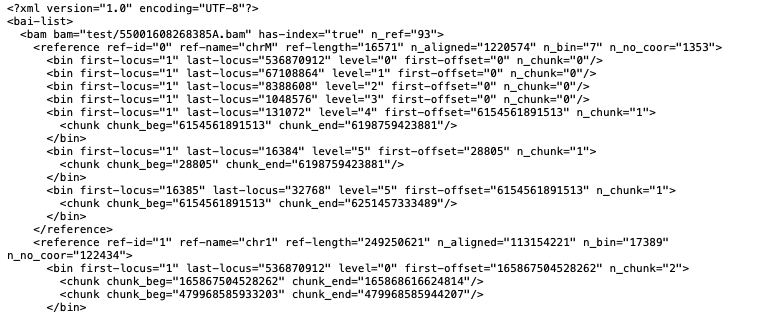
Because the bai file itself does not include the chromosomes or their lengths, we pull that information from IGB (which is what allows us to view "just" the bai file). However, this makes two assumptions. That the user aligned their reads to that specific version of the genome (i.e. against hg38), and that when the bam file was sorted the chromosomes were sorted into the same order in which they appear in IGB (or the genome.txt). When the bai file is read, we can see when we transition from one chromosome to the next, but we cannot tell what chromosome it is. So if the chromosomes are sorted in a different order in the user's bai file, there is no way for us to know that. For example, the attached file (55001608268385A) looked odd to me was because the mitochondrial chromosome was at the very beginning of the file instead of chromosome 1 (see attached screenshots), creating a kind of off by one error for the chromosomes.
The assumption then becomes that the genome.txt and the files bai are in the same order. However, the genome.txt is not necessarily sorted lexicographically, as we will manually rearrange the chromosomes to make more sense to the user (i.e. chr1,chr2,chrX,anotherSmallChromosome). In addition, I am not sure that the bai files themselves are sorted lexicographically by samtools. When I tested samtools, the order that the chromosomes appeared in the .bam/.bai was determined by the order of the chromosomes within the bam header. Rearranging the chromosomes in the bam header then sorting the file rearranged the reads into the same order as the header. Therefore, we cannot assume there is a default sort order even if the bam/bai files have been sorted with samtools.
Because we cannot assume there is a sort order, I would recommend altering the index hacking logic.
1. When the user loads a bai file, check to see if there is a bam file that we can read the header from. Use the header to determine the order of the chromosomes.
2. If no bam file can be found, compare the bin sizes to the chromosome sizes and use that to order the chromosomes. Level 5 bins are 16kbp, which should be relatively accurate for larger chromosomes.