Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:5
-
Epic Link:
-
Sprint:Spring 3, Spring 4, Spring 5, Spring 6, Spring 7
Description
Data has been submitted to SRA and now needs to be reviewed. To do this we perform a rerun of the data by pulling it from SRA directly and pushing it through the nextflow pipeline and prepping it for IGB quick load. To confirm that the data on SRA is correct we can make a few comparisons with the original data.
- Compare files sizes of the original data to the new rerun file sizes.
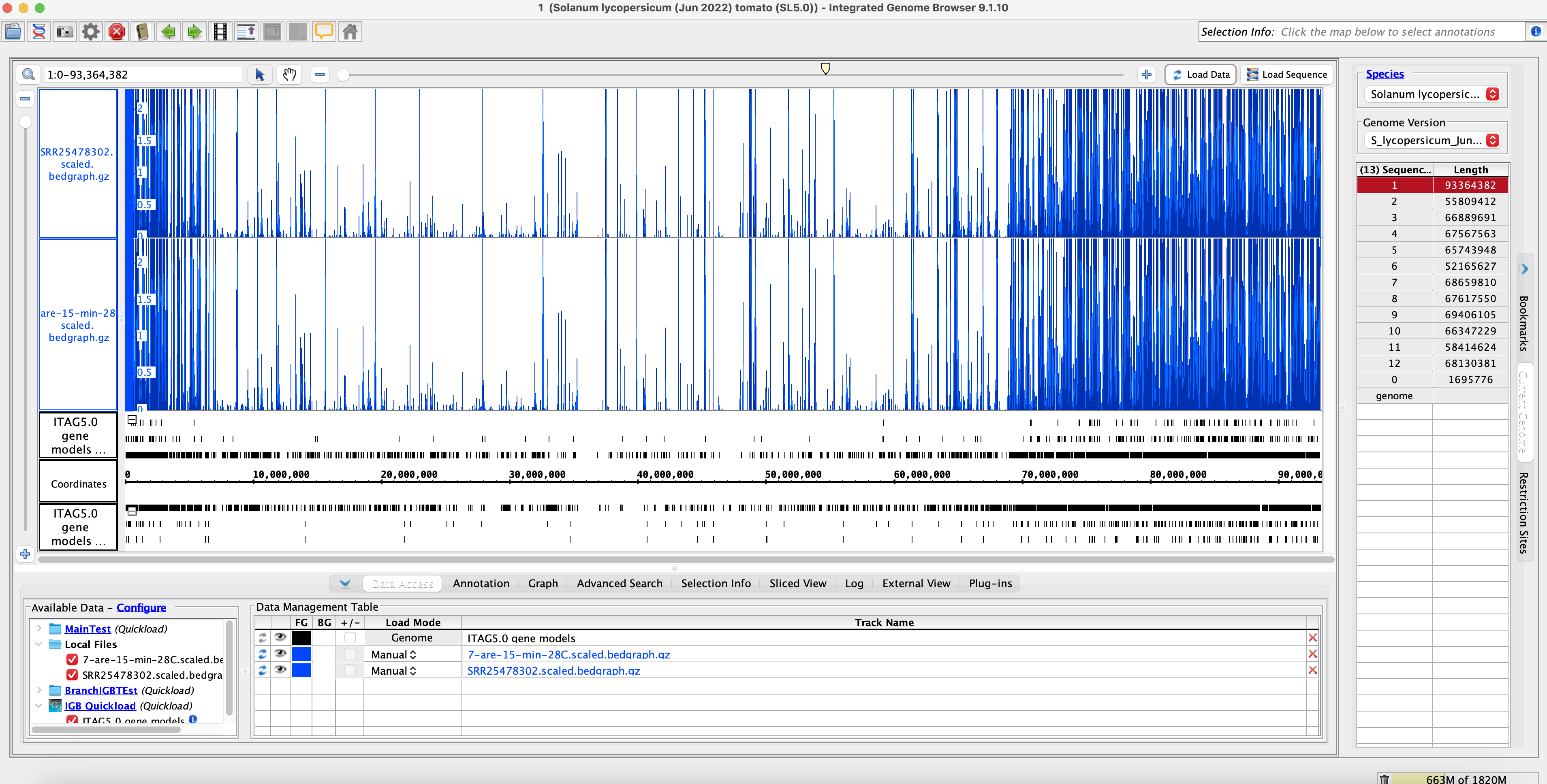
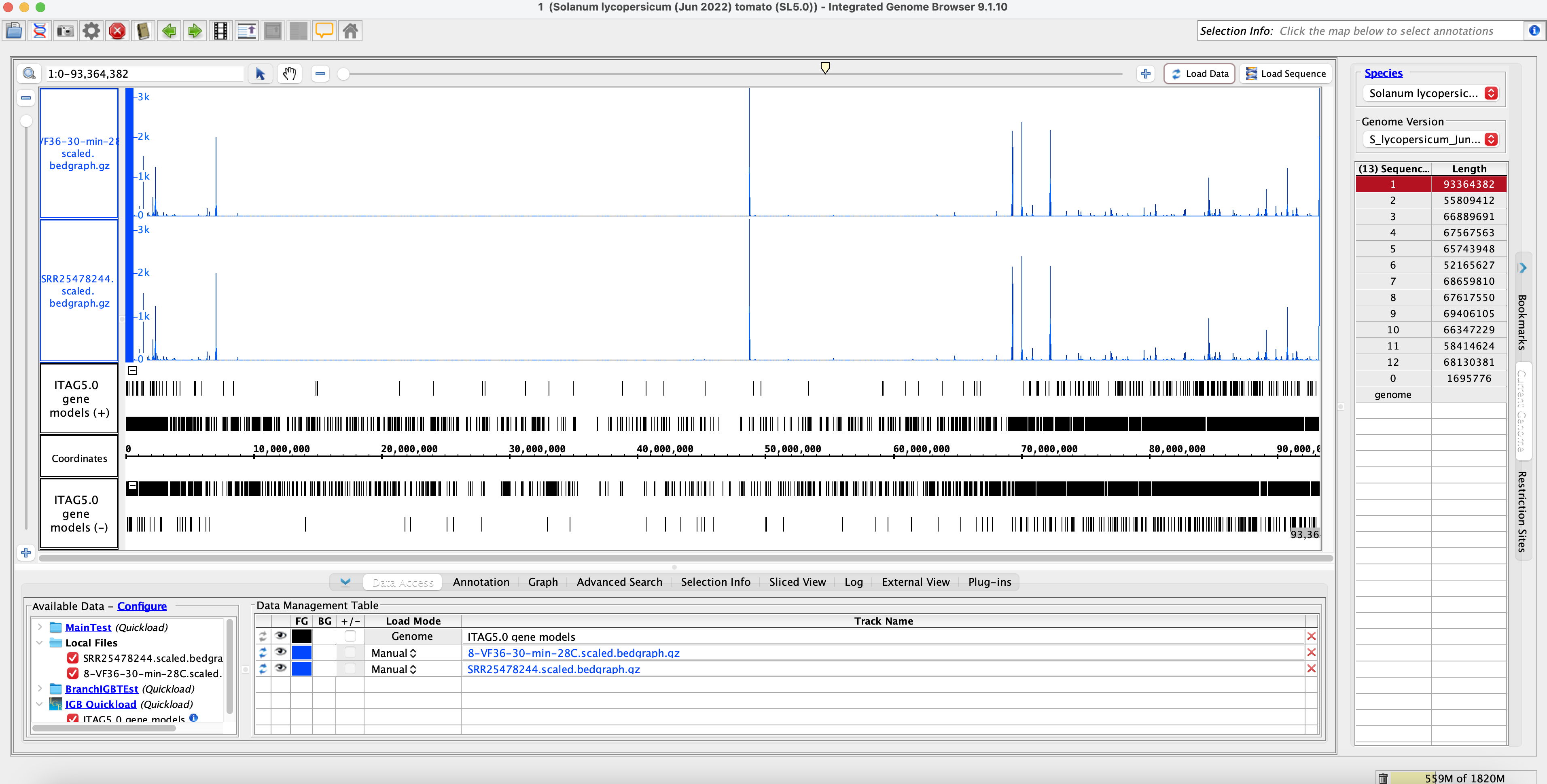
- Open IGB and compare bam and coverage graphs with the original and rerun data.
- Perform these comparisons for SL4 and SL5
Muday cluster Directories:
Original: /projects/tomato_genome/fnb/dataprocessing/muday-144
Re-run: /projects/tomato_genome/fnb/dataprocessing/SRP460750
Attachments




Issue Links
- relates to
-
-
- Closed
-
-
-
- Closed
-
-
-
- Closed
-
-
-
- To-Do
-
-
-
- Closed
-
Activity
Checking the first 20 sequences of each file
The next step is this:
- Zcat each fastq file, and awk to capture every 4th line (the actual sequence line). Head this result to get just the 20 lines of sequence. Write these results to a new file. This will act as a fingerprint (150 characters * 5) (( 20 lines will be 5 sequences total, still plenty for a finger print))
- Compare each of the pre and post fingerprints and summarize into table. Compare to Ann's results above, should match up.
- And then plan on changing SRA accordingly.
Oh this all fails if the SRA changes the order of the sequences or the composition in any way!! Which would be horrifying.
Awk command to catch the 2nd line of the fastq
This will take 20 lines from the zipped fastq, and print out JUST the 2nd line (aka the sequence).
zcat A.28.45.9_R2.fastq.gz | head -n 20 | awk ' NR%4==2
{ print $1 }'
With this we make our fingerprints.
Folder for COMPARING fastq pre and post:
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra
Fingerprints for Pre fastq files:
Fingerprints for Post fastq files:
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra/post-srafiles
The 1 line command to make fingerprints:
PRE
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra/post-srafiles$
while read file; do echo $
.fastq.gz | head -n 20 | awk ' NR%4==2
{print $1}' > ./pre-finalfiles/${file}-5lines.txt ; done < prenames.txtPOST
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra/post-srafiles$
while read file; do echo ${file}; zcat /projects/tomato_genome/fnb/dataprocessing/SRP460750/nfcore-SL4/${file}.fastq.gz | head -n 20 | awk ' NR%4==2 {print $1}
' > ./post-srafiles/$
{file}-5lines.txt ; done < postsrafiles.txt
Fingerprints complete.
Now to compare them!
We need to iterate through each PRE fingerprint file and find its matching partner in POST sra.
This is now done.
Summarizing the above:
- We think the sequence data itself is fine because the pre- and post-SRA submission nf-core/rnaseq profiles were identical
- However, we found that 16 of the SRR entries had the wrong sample codes.
- We identified the affected files using two methods - (1) comparing multiqc-generated General Statistics profiles (see Ann's R code) and (2) comparing the first 20 lines from the sequence data files (see Rob's comments above).
- In addition, we identified the files that need to have their sample codes re-assigned, and how. A csv file reporting these is in the "results" folder of the git repository. See above comments.
Now that we know which samples were affected and how to re-label them in the SRA, we can close this ticket.
Download Data from Cluster:
Original Muday Bam files:
Rerun Muday Bam files:
Original Muday Scaled Coverage Graphs:
Rerun Muday Scaled Coverage Graphs:
Notes: Had to compress bam files in each directory before downloading. I also have to use 'Muday-lab-RNA-samples-for-sample-name-conversion' file from flavonoid repo to check the correct names of the samples due to mistakes in the lab which led to sample switching.