Details
-
Type:
Task
-
Status: Closed (View Workflow)
-
Priority:
 Major
Major
-
Resolution: Done
-
Affects Version/s: None
-
Fix Version/s: None
-
Labels:None
-
Story Points:5
-
Epic Link:
-
Sprint:Spring 3, Spring 4, Spring 5, Spring 6, Spring 7
Description
Data has been submitted to SRA and now needs to be reviewed. To do this we perform a rerun of the data by pulling it from SRA directly and pushing it through the nextflow pipeline and prepping it for IGB quick load. To confirm that the data on SRA is correct we can make a few comparisons with the original data.
- Compare files sizes of the original data to the new rerun file sizes.
- Open IGB and compare bam and coverage graphs with the original and rerun data.
- Perform these comparisons for SL4 and SL5
Muday cluster Directories:
Original: /projects/tomato_genome/fnb/dataprocessing/muday-144
Re-run: /projects/tomato_genome/fnb/dataprocessing/SRP460750
Attachments




Issue Links
- relates to
-
-
- Closed
-
-
-
- Closed
-
-
-
- Closed
-
-
-
- To-Do
-
-
-
- Closed
-
Activity
Next step: Use multiqc reports to sanity check the sample switch. Product of this work would be a visualization/picture that would allow other people to reach a similar conclusion that the SRA submissions were accurate. Also that the sample switching was accurate.
- Method 1: Use the multiqc reports from the original data and compare it with the report for the SRA rerun data. Make conclusions whether the data are the same and why with visuals from the reports. Do this with a sample that didn't have sample switching and one that did.
- Method 2: Use IGB to show coverage graphs of one chromosome for original and rerun. Do this twice, one without sample switching names and one with sample switching. Screenshot the comparisons.
Summary
Goal: Now that the original flavonoid muday-144 data has been submitted to SRA and rerun with nextflow, we can compare the original data to the SRA data to make sure everything was submitted correctly. To check this we want to compare the original data to the SRA data to ensure consistency. Comparisons will be made with multiqc reports and IGB visualizations of comparing coverage graphs. As we know there was a sample switching issue that was resolved. But during this comparison I will compare a sample that had no sample switching and one that did to double check the names.
Method 1: MultiQc reports SL5 Comparisons:
No sample Switch name = SRR25478302 = A.28.15.7 = 7-are-15-min-28C (Cluster Name)


Sample switch name = SRR25478244 = V.28.30.8 (Original Name) = A.28.15.8 (Actual Name) = 8-VF36-30-min-28C (Cluster Name)


Conclusion of Method 1:
- The No sample Switch name is accurate and have the same exact nextflow mapping results. In other words, the original sample and the SRA rerun sample have the exact same results in the multiqc report after being run with nextflow. The sample switch name is accurate with the same exact results and the fix to the naming is correct. In other words, by using the corrected name we can confirm with the report that there was a switching issue and the names are now fixed. The SRA submission in summary is the same as the original data but now with the fixed names.
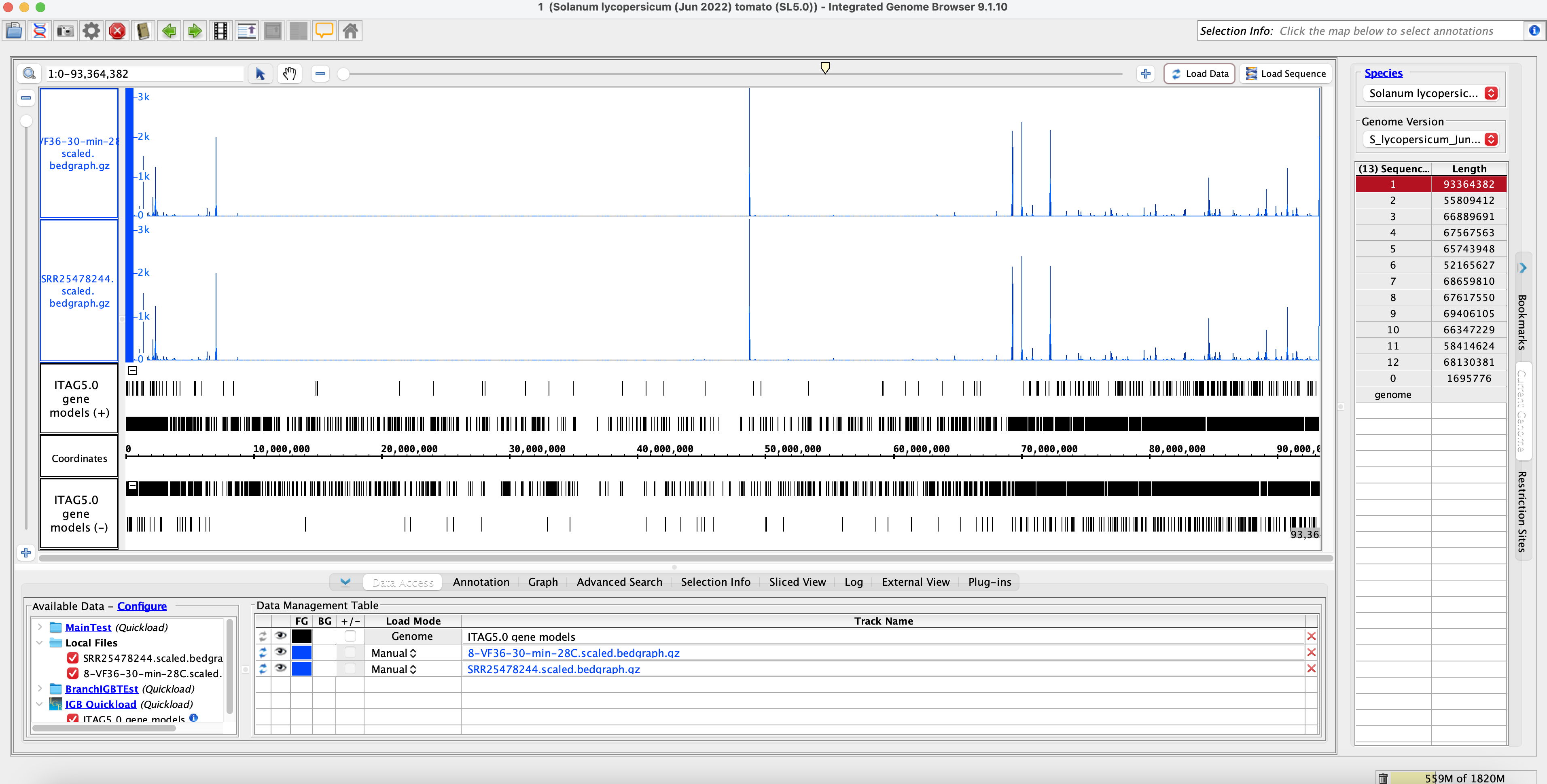
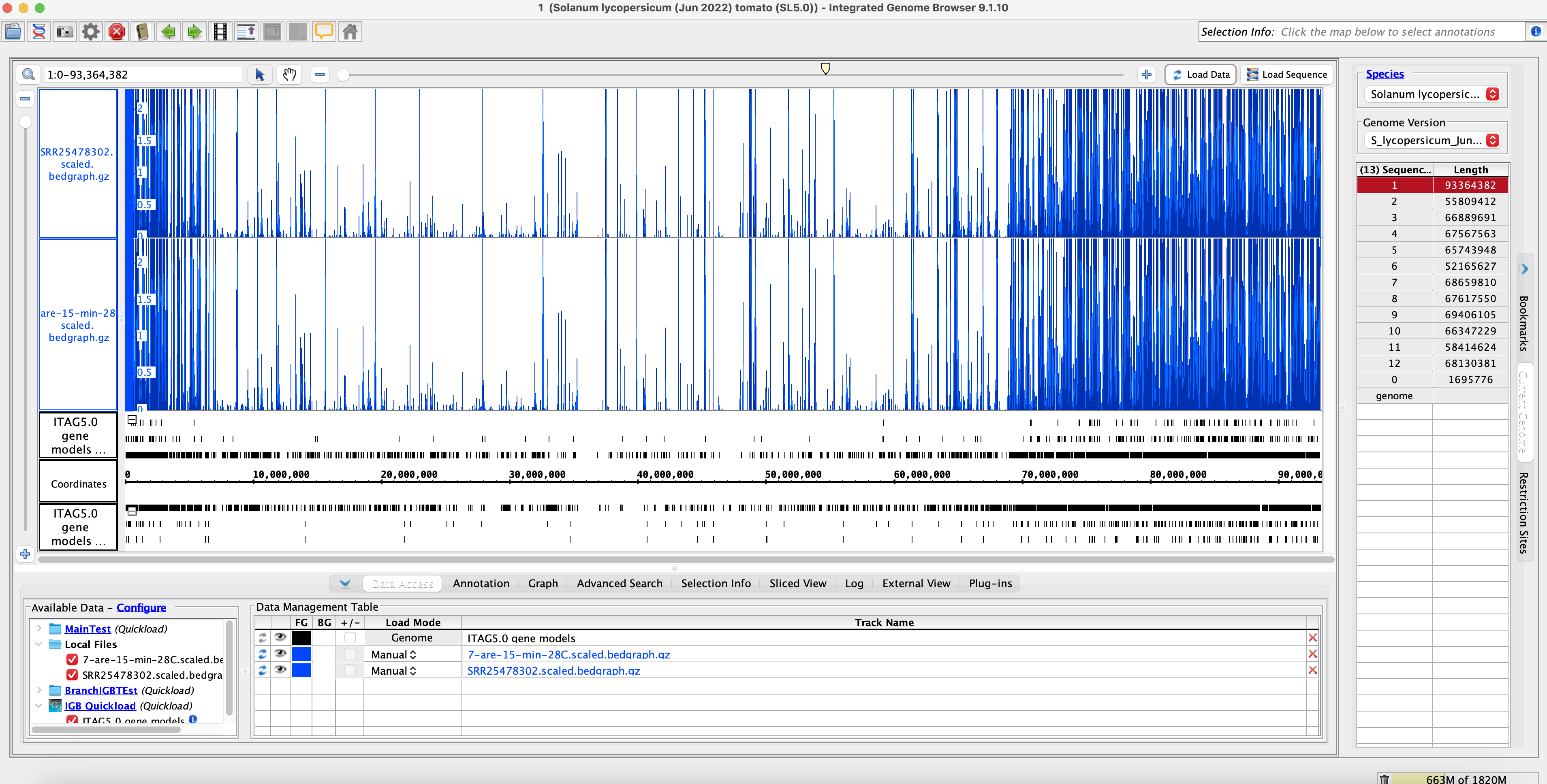
Method 2: IGB Coverage Graphs SL5 Comparisons
No sample Switch name = SRR25478302 = A.28.15.7 = 7-are-15-min-28C (Cluster Name)
Sample switch name = SRR25478244 = V.28.30.8 (Original Name) = A.28.15.8 (Actual Name) = 8-VF36-30-min-28C (Cluster Name)
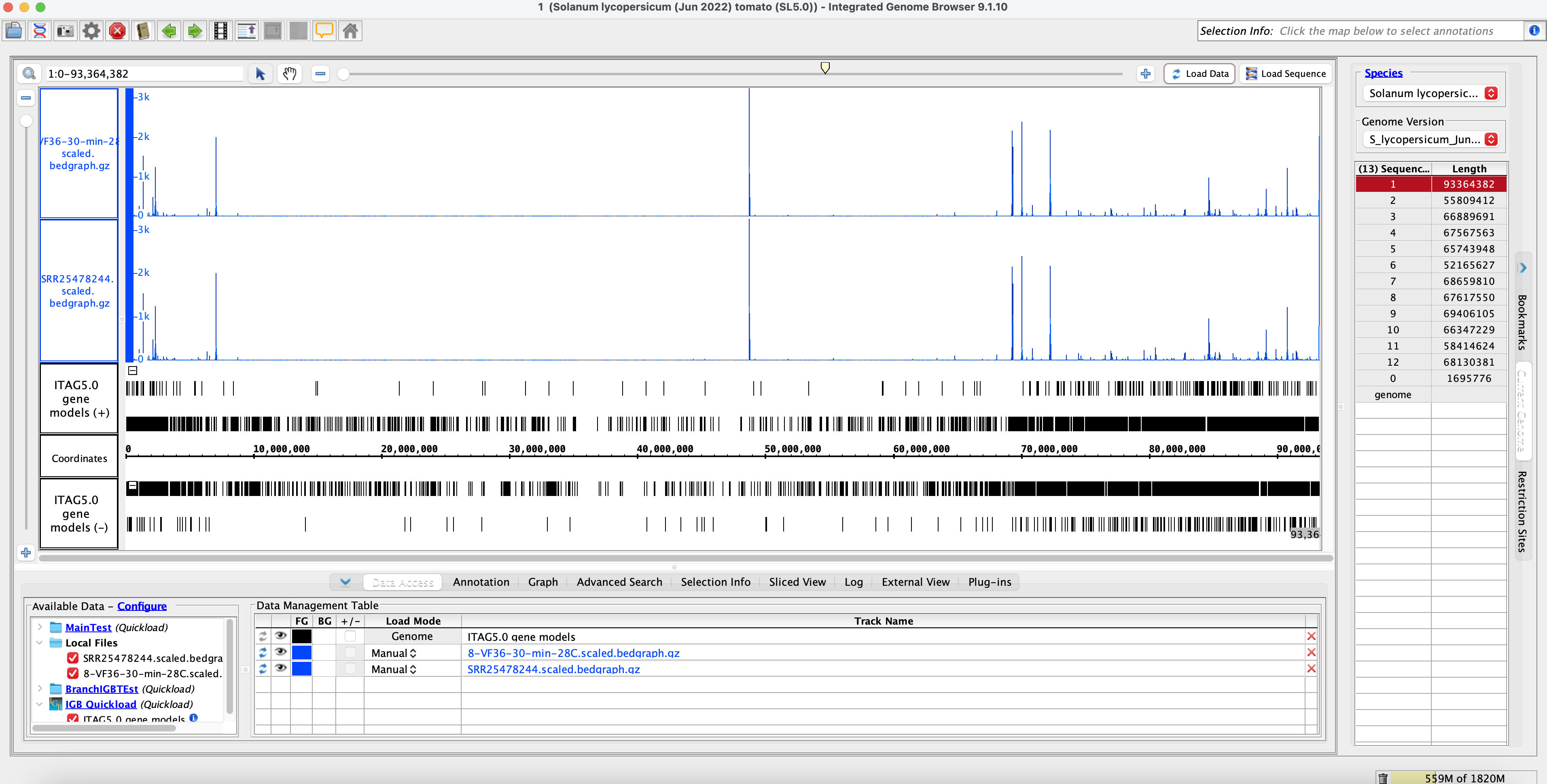
Conclusion of Method 2:
- Visually we can see that the data is similar. The coverage graphs are the same, indicating that the original data matches with the SRA submission data. The sample name was also switched to the correct sample name.
Questions and Review:
- Do we think these methods achieve our goal of comparing original data to SRA rerun data?
- Should we perform these methods for every single sample in the experiment or are these samples enough proof?
- Once we can confirm that the new SRA submission is accurate should we move the original data from the cluster to an off sight location like 'Glacier'?
- Should we show muday lab these comparisons or go ahead and submit the SRA data to quick load and then show them the new SRA data that is available?
Next step: Create sample sheet for SRA data and submit to quick load.
This all looks good to me and we should charge forward!
But Let's get Ann's thoughts first!
I liked the visualization from the multi qc report. The images from nextflow runs of the same sequence data before and after submission to SRA were the same.
Based on this, I think we should now compare all the samples in exactly this way. If there is a problem with any of the samples, we will likely find out immediately.
My recommendation for the next step:
I suggest that Molly Davis create a ticket to use the "compare images from multiqc" on all the SRA files for the Muday flavonoid data set. To make it easier to look at all the data, she could create a Power Point document with the images shown side-by-side - "pre" and "post" SRA-submission.
Once the PowerPoint is made, we could add it to the BitBucket repository as documentation that this QA set was completed.
Once this new ticket is made and added to the board (current sprint is fine) let's move this ticket to DONE.
Thank you Molly Davis and Robert Reid !!!!
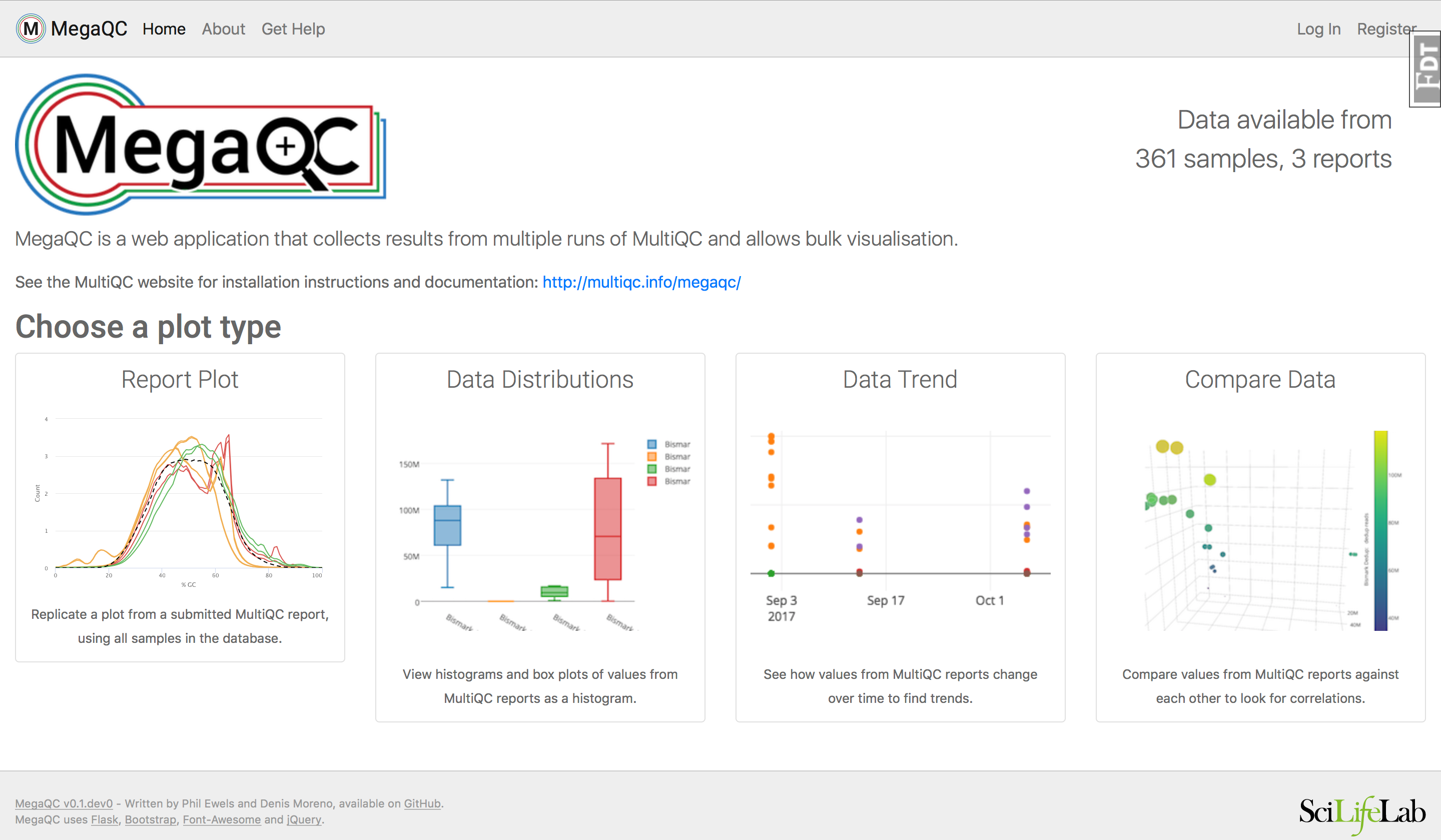
As of right now comparing many samples from two mutliQC report seems like it would take a lot of time. I found this tool MegaQC which allows you to look at and compare many multiQC reports at once:
https://megaqc.info/docs/index.html
https://github.com/MultiQC/MegaQC?tab=readme-ov-file
MegaQC is a web application that you can install and run on your own network. It collects and visualises data parsed by MultiQC across multiple runs.
Other options: https://multiqc.info/docs/usage/downstream/
Chronqc:
https://chronqc.readthedocs.io/en/latest/run_chronqc.html#generating-chronqc-plots
Just a quick comment on above:
The main (only) goal here is: Show that the original data and the SRA-downloaded data are the same.
Obviously, since there are millions (lots and lots!) of sequence reads per sample, we can't compare each one.
However, the "horizontal strip" images you showed in a previous comment (e.g., 7-are-15-min-28C-multiqcreport.png) really seemed to have the perfect amount of detail required to compare samples before and after upload to the SRA.
Indeed, that image looked like a kind of summary profile of the much bigger data set as it behaved when we aligned it to a reference.
How about making a google slidedeck:
https://docs.google.com/presentation/d/17NeX_extZ6pTOBlImOs2-Bp5V6wNZP_O5KSCCTu5t5g/edit?usp=sharing
Check Method:
The method of comparing mutliQC reports has been useful and accurate. It actually found some original file names that were not converted correctly. To check that this method is sound, Dr. Reid and I decided it would be best to compare the original sequence files (FASTQ) on the cluster. These comparisons were made with the following files locations and scripts:
Original fastq file location: /projects/tomato_genome/rnaseq/muday144-timeSeries-checkReadMEFIRST/00_fastq
SRA fastq files location: /projects/tomato_genome/fnb/dataprocessing/SRP460750/nfcore-SL5
Sequence information script:
for file in *gz; do /projects/tomato_genome/sw/sequence_info -n $file >> seqsummary.txt; done
The goal of comparing the original sequences is to confirm that the files were not matched up to the corrected sample names for some of the samples.
Loraine update:
- I wrote a Markdown to compare General Statistics pre- and post-submission.
- Preliminary results (assuming no bugs in my code) is that a lot of samples look like they got mixed up somehow
See files:
- 72_F3H_PollenTube/Documentation/CheckSRA-SL5.xlsx
- 72_F3H_PollenTube/CheckSRA.Rmd
- 72_F3H_PollenTube/CheckSRA.html
in https://bitbucket.org/hotpollen/flavonoid-rnaseq
Next steps: Check for bugs in my code.
I have added testing code that double-checks what I've been assuming are the correct sample codes for each original file name using the Muday lab spreadsheet mentioned above.
My code appears correct.
I have pushed a new version to the repository.
Moving this forward to review. Also, we have re-opened and linked this ticket to the original SRA submission ticket to make it easier for Robert Reid to review this work and determine the best way to update the SRA records or re-submit things as needed.
Lastly, I added some new code that suggests new sample codes be assigned to 16 sample records.
The suggested new sample codes are in a new file "updateSRA.csv"
See:
- 72_F3H_PollenTube/results/updateSRA.csv
in https://bitbucket.org/hotpollen/flavonoid-rnaseq "main" branch.
attn: Robert Reid and Molly Davis
Confirming that the Fastq file headers get changed after SRA submission is true.
In this folder I have summaries of the MD5 results.
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/final-files
I did the MD5 of the zipped fastq files prior to them being loaded to the SRA.
I also did the MD5 of the SRA post downloaded files which can be found here on the HPC:
/projects/tomato_genome/fnb/dataprocessing/SRP460750/nfcore-SL4
Comparing he MD5 codes after sorting, there are no overlaps of ANY of the 144 files.
Running the MD5 checks
/projects/tomato_genome/fnb/dataprocessing/SRP460750/nfcore-SL4
for file in *gz; do echo $file; md5sum $file >> /projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/final-files/postdownloaded-md5.txt; done
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/final-files$
for file in *gz; do echo $file; md5sum $file >> md5.txt; done
Checking the first 20 sequences of each file
The next step is this:
- Zcat each fastq file, and awk to capture every 4th line (the actual sequence line). Head this result to get just the 20 lines of sequence. Write these results to a new file. This will act as a fingerprint (150 characters * 5) (( 20 lines will be 5 sequences total, still plenty for a finger print))
- Compare each of the pre and post fingerprints and summarize into table. Compare to Ann's results above, should match up.
- And then plan on changing SRA accordingly.
Oh this all fails if the SRA changes the order of the sequences or the composition in any way!! Which would be horrifying.
Awk command to catch the 2nd line of the fastq
This will take 20 lines from the zipped fastq, and print out JUST the 2nd line (aka the sequence).
zcat A.28.45.9_R2.fastq.gz | head -n 20 | awk ' NR%4==2
{ print $1 }'
With this we make our fingerprints.
Folder for COMPARING fastq pre and post:
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra
Fingerprints for Pre fastq files:
Fingerprints for Post fastq files:
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra/post-srafiles
The 1 line command to make fingerprints:
PRE
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra/post-srafiles$
while read file; do echo $
.fastq.gz | head -n 20 | awk ' NR%4==2
{print $1}' > ./pre-finalfiles/${file}-5lines.txt ; done < prenames.txtPOST
/projects/tomato_genome/rnaseq/renamed_MudayTimeCourseSequences/troubleshoot-sra/post-srafiles$
while read file; do echo ${file}; zcat /projects/tomato_genome/fnb/dataprocessing/SRP460750/nfcore-SL4/${file}.fastq.gz | head -n 20 | awk ' NR%4==2 {print $1}
' > ./post-srafiles/$
{file}-5lines.txt ; done < postsrafiles.txt
Fingerprints complete.
Now to compare them!
We need to iterate through each PRE fingerprint file and find its matching partner in POST sra.
This is now done.
Summarizing the above:
- We think the sequence data itself is fine because the pre- and post-SRA submission nf-core/rnaseq profiles were identical
- However, we found that 16 of the SRR entries had the wrong sample codes.
- We identified the affected files using two methods - (1) comparing multiqc-generated General Statistics profiles (see Ann's R code) and (2) comparing the first 20 lines from the sequence data files (see Rob's comments above).
- In addition, we identified the files that need to have their sample codes re-assigned, and how. A csv file reporting these is in the "results" folder of the git repository. See above comments.
Now that we know which samples were affected and how to re-label them in the SRA, we can close this ticket.
Download Data from Cluster:
Original Muday Bam files:
Rerun Muday Bam files:
Original Muday Scaled Coverage Graphs:
Rerun Muday Scaled Coverage Graphs:
Notes: Had to compress bam files in each directory before downloading. I also have to use 'Muday-lab-RNA-samples-for-sample-name-conversion' file from flavonoid repo to check the correct names of the samples due to mistakes in the lab which led to sample switching.